mLLMCelltype
mLLMCelltype 是一款专为单细胞 RNA 测序(scRNA-seq)数据设计的自动化细胞类型注释工具。在单细胞分析中,准确识别细胞类型往往依赖专家经验或参考数据集,不仅耗时且容易受单一模型偏差影响。mLLMCelltype 创新性地引入了“多大型语言模型共识”机制,通过整合 OpenAI、Claude、Gemini、DeepSeek 等十余种主流大模型的预测结果,让多个 AI“专家”对同一数据进行多轮讨论与交叉验证,从而达成更可靠的注释结论。
该方法无需依赖外部参考数据集,即可实现高达 95% 的注释准确率,并能输出不确定性指标(如共识比例和香农熵),帮助研究者识别存疑结果。它无缝兼容 Scanpy 和 Seurat 等常用生物信息学流程,既提供 Python/R 代码包供开发者调用,也拥有免安装的网页版界面,极大降低了使用门槛。
mLLMCelltype 特别适合生物信息学研究人员、基因组学科学家以及需要处理单细胞数据的实验室团队使用。其独特的多模型迭代讨论架构,有效克服了单一 AI 模型的局限性,为复杂生物数据的解读提供了更加稳健、透明的解决方案。
使用场景
某生物信息学研究员正在分析一项罕见的神经退行性疾病单细胞测序数据,急需在缺乏高质量参考图谱的情况下,准确识别样本中未知的稀有免疫细胞亚群。
没有 mLLMCelltype 时
- 依赖单一模型导致偏差:仅使用单个大模型进行注释时,容易受该模型训练数据偏见影响,将新型细胞错误归类为常见类型。
- 人工复核耗时巨大:面对模型给出的模糊预测,研究员需手动查阅大量文献逐一验证基因标记物,耗费数天时间。
- 缺乏置信度评估:传统方法难以量化注释的不确定性,导致研究人员无法判断哪些细胞类型的鉴定结果是可靠的,哪些存疑。
- 冷启动困难:由于缺乏匹配的参考数据集,基于映射的传统工具完全失效,分析工作陷入停滞。
使用 mLLMCelltype 后
- 多模型共识消除偏见:mLLMCelltype 调用 GPT-5.2、Claude-4.6 等十余个大模型进行“集体会诊”,通过共识机制有效纠正了单一模型的误判。
- 自动化迭代讨论:工具内置的多轮讨论机制自动评估基因表达证据并优化注释,将原本数天的人工复核工作缩短至几小时。
- 明确的不确定性指标:系统直接输出香农熵和共识比例,帮助研究员快速定位那些需要重点关注的低置信度细胞群。
- 无需参考数据即可运行:凭借强大的生成式能力,mLLMCelltype 直接在无参考图谱条件下完成了高精度(达 95%)的细胞类型鉴定。
mLLMCelltype 通过整合多模型智慧与量化不确定性,将单细胞注释从“盲目猜测”转变为可信赖的自动化科学发现流程。
运行环境要求
- Linux
- macOS
- Windows
未说明 (该工具通过 API 调用云端大模型,本地无需 GPU)
未说明
快速开始






mLLMCelltype: 多大语言模型共识框架用于细胞类型注释
mLLMCelltype 是一个基于多大语言模型共识的自动化单细胞RNA测序(scRNA-seq)数据细胞类型注释框架。该框架整合了包括 OpenAI GPT-5.2、Anthropic Claude-4.6/4.5、Google Gemini-3、X.AI Grok-4、DeepSeek-V3、Alibaba Qwen3、Zhipu GLM-4、MiniMax、Stepfun 和 OpenRouter 在内的多个大型语言模型,通过基于共识的预测来提高注释准确性。
摘要
mLLMCelltype 是一款开源的单细胞转录组学分析工具,利用多个大型语言模型从基因表达数据中识别细胞类型。该软件采用共识方法,即多个模型对同一数据进行分析并将它们的预测结果进行综合,从而有助于减少误差并提供不确定性度量。这种方法通过整合多个模型的预测结果,相较于单一模型方法具有显著优势。mLLMCelltype 可与 Scanpy 和 Seurat 等单细胞分析平台集成,使研究人员能够将其纳入现有的工作流程中。该方法无需参考数据集即可进行注释。
在我们的基准测试中(Yang 等,2025),共识方法在测试数据集上达到了高达95%的准确率。
目录
Web 应用程序:浏览器端界面可在 mllmcelltype.com 上访问(无需安装)。
另请参阅:FlashDeconv — 用于空间转录组学(Visium、Visium HD、Stereo-seq)的细胞类型去卷积工具。
主要特性
- 多大语言模型共识:整合多个大语言模型的预测结果,以减少单一模型的局限性和偏差
- 模型支持:兼容 OpenAI、Anthropic、Google 等10余家大语言模型提供商
- 迭代讨论:大语言模型通过多轮讨论评估证据并优化注释
- 不确定性量化:提供共识比例和香农熵指标,以识别不确定的注释
- 跨模型验证:通过多模型比较减少错误预测
- 抗噪能力:即使标记基因列表不完善也能保持较高的准确性
- 层次化注释:支持多分辨率分析,并进行一致性检查
- 无参考依赖:无需预训练或参考数据集即可完成注释
- 文档记录:完整记录推理过程,确保透明性
- 集成性:与 Scanpy/Seurat 工作流及标记基因输出兼容
- 可扩展性:随着新大语言模型的出现,可随时添加支持
有关更改日志和更新,请参阅 NEWS.md。
安装
R 版本
# 从 CRAN 安装(推荐)
install.packages("mLLMCelltype")
# 或从 GitHub 安装开发版本
devtools::install_github("cafferychen777/mLLMCelltype", subdir = "R")
Python 版本
![]()
快速入门:无需任何安装,即可在 Google Colab 中试用 mLLMCelltype。点击上方徽章即可打开包含示例和分步指导的交互式笔记本。
# 从 PyPI 安装
pip install mllmcelltype
# 或从 GitHub 安装(注意子目录参数)
pip install git+https://github.com/cafferychen777/mLLMCelltype.git#subdirectory=python
关于依赖的重要说明
mLLMCelltype 采用模块化设计,不同大语言模型提供商的库为可选依赖。根据您计划使用的模型类型,需要安装相应的包:
# 使用 OpenAI 模型(GPT-5 等)
pip install "mllmcelltype[openai]"
# 使用 Anthropic 模型(Claude)
pip install "mllmcelltype[anthropic]"
# 使用 Google 模型(Gemini)
pip install "mllmcelltype[gemini]"
# 一次性安装所有可选依赖
pip install "mllmcelltype[all]"
如果您遇到类似 ImportError: cannot import name 'genai' from 'google' 的错误,说明您需要安装对应的提供商包。例如:
# 对于 Google Gemini 模型
pip install google-genai
支持的模型
- OpenAI: GPT-5.2/GPT-5/GPT-4.1(API密钥)
- Anthropic: Claude-4.6-Opus/Claude-4.5-Sonnet/Claude-4.5-Haiku(API密钥)
- Google: Gemini-3-Pro/Gemini-3-Flash(API密钥)
- 阿里巴巴: Qwen3-Max(API密钥)
- DeepSeek: DeepSeek-V3/DeepSeek-R1(API密钥)
- Minimax: MiniMax-M2.1(API密钥)
- Stepfun: Step-3(API密钥)
- 智谱: GLM-4.7/GLM-4-Plus(API密钥)
- X.AI: Grok-4/Grok-3(API密钥)
- OpenRouter: 通过单一API访问多种模型(API密钥)
- 支持来自OpenAI、Anthropic、Meta、Google、Mistral等公司的模型
- 格式:'provider/model-name'(例如,'openai/gpt-5.2'、'anthropic/claude-opus-4.5')
- 提供带有
:free后缀的免费模型(例如,'deepseek/deepseek-r1:free'、'meta-llama/llama-4-maverick:free') - 注意:免费套餐限制为每天50次请求(充值10美元以上可提升至每天1000次),每分钟20次请求。部分模型可能不可用。
使用示例
Python
# 使用mLLMCelltype结合Scanpy进行单细胞RNA测序细胞类型注释的示例
import scanpy as sc
import pandas as pd
from mllmcelltype import annotate_clusters, interactive_consensus_annotation
import os
# 注意:导入mllmcelltype时会自动配置日志记录
# 如有需要,可使用logging模块自定义日志设置
# 加载您的AnnData格式的单细胞RNA测序数据集
adata = sc.read_h5ad('your_data.h5ad') # 替换为您自己的scRNA-seq数据集路径
# 如果尚未完成,执行Leiden聚类以识别细胞群体
if 'leiden' not in adata.obs.columns:
print("正在计算Leiden聚类以识别细胞群体...")
# 预处理单细胞数据:归一化计数并进行对数转换,用于基因表达分析
if 'log1p' not in adata.uns:
sc.pp.normalize_total(adata, target_sum=1e4) # 归一化至每细胞10,000个计数
sc.pp.log1p(adata) # 对归一化后的计数取自然对数
# 降维:计算scRNA-seq数据的主成分分析
if 'X_pca' not in adata.obsm:
sc.pp.highly_variable_genes(adata, min_mean=0.0125, max_mean=3, min_disp=0.5) # 选择信息量丰富的基因
sc.pp.pca(adata, use_highly_variable=True) # 计算主成分
# 细胞聚类:构建KNN图并执行Leiden社区检测
sc.pp.neighbors(adata, n_neighbors=10, n_pcs=30) # 构建用于聚类的KNN图
sc.tl.leiden(adata, resolution=0.8) # 使用Leiden算法识别细胞群体
print(f"Leiden聚类已完成,共识别出{len(adata.obs['leiden'].cat.categories)}个不同的细胞群体")
# 利用差异表达分析鉴定每个细胞簇的标记基因
sc.tl.rank_genes_groups(adata, 'leiden', method='wilcoxon') # 使用Wilcoxon秩和检验检测标记基因
# 提取每个细胞簇的 top 10 标记基因,用于细胞类型注释
marker_genes = {}
for i in range(len(adata.obs['leiden'].cat.categories)):
# 选择每个簇中差异表达最显著的前10个基因作为标记
genes = [adata.uns['rank_genes_groups']['names'][str(i)][j] for j in range(10)]
marker_genes[str(i)] = genes
# 重要提示:mLLMCelltype要求使用基因符号(如KCNJ8、PDGFRA),而非Ensembl ID(如ENSG00000176771)
# 如果您的AnnData对象使用的是Ensembl ID,请将其转换为基因符号以便准确注释:
# 示例转换代码:
# if 'Gene' in adata.var.columns: # 检查元数据中是否已有基因符号
# gene_name_dict = dict(zip(adata.var_names, adata.var['Gene']))
# marker_genes = {cluster: [gene_name_dict.get(gene_id, gene_id) for gene_id in genes]
# for cluster, genes in marker_genes.items()}
# 重要提示:mLLMCelltype要求使用数值型簇ID
# “cluster”列必须包含数值或可转换为数值的值。
# 非数值型簇ID(如“cluster_1”、“T_cells”、“7_0”)可能导致错误或异常行为。
# 如果您的数据包含非数值型簇ID,需创建原始ID与数值ID之间的映射:
# 示例标准化代码:
# original_ids = list(marker_genes.keys())
# id_mapping = {original: idx for idx, original in enumerate(original_ids)}
# marker_genes = {str(id_mapping[cluster]): genes for cluster, genes in marker_genes.items()}
# 配置用于共识注释的大型语言模型API密钥
# 至少需要一个API密钥才能进行多LLM共识注释
os.environ["OPENAI_API_KEY"] = "your-openai-api-key" # 用于GPT-5.2/5/4.1模型
os.environ["ANTHROPIC_API_KEY"] = "your-anthropic-api-key" # 用于Claude-4.6/4.5模型
os.environ["GEMINI_API_KEY"] = "your-gemini-api-key" # 用于Google Gemini-3模型
os.environ["QWEN_API_KEY"] = "your-qwen-api-key" # 用于阿里巴巴Qwen3模型
# 其他可选的LLM提供商,以增强共识多样性:
# os.environ["DEEPSEEK_API_KEY"] = "your-deepseek-api-key" # 用于DeepSeek-V3模型
# os.environ["ZHIPU_API_KEY"] = "your-zhipu-api-key" # 用于智谱GLM-4模型
# os.environ["STEPFUN_API_KEY"] = "your-stepfun-api-key" # 用于Stepfun模型
# os.environ["MINIMAX_API_KEY"] = "your-minimax-api-key" # 用于MiniMax模型
# os.environ["OPENROUTER_API_KEY"] = "your-openrouter-api-key" # 用于通过OpenRouter访问多种模型
# 执行多LLM共识细胞类型注释,并进行迭代讨论
consensus_results = interactive_consensus_annotation(
marker_genes=marker_genes, # 每个簇的标记基因字典
species="human", # 指定生物种类,以便进行适当的细胞类型注释
tissue="blood", # 指定组织背景,以提高注释准确性
models=["gpt-5.2", "claude-sonnet-4-5-20250929", "gemini-3-pro", "qwen3-max"], # 多个LLM用于共识
consensus_threshold=1, # 达成共识所需的最低比例
max_discussion_rounds=3 # 模型之间进行细化讨论的轮次数
)
# 或者,使用OpenRouter通过单一API访问多种模型
# 这对于访问带有`:free`后缀的免费模型尤其有用
os.environ["OPENROUTER_API_KEY"] = "your-openrouter-api-key"
# 使用免费的 OpenRouter 模型示例(无需积分)
free_models_results = interactive_consensus_annotation(
marker_genes=marker_genes,
species="human",
tissue="blood",
models=[
{"provider": "openrouter", "model": "meta-llama/llama-4-maverick:free"}, # Meta Llama 4 Maverick(免费)
{"provider": "openrouter", "model": "venice/uncensored:free"}, # Venice Uncensored(免费)
{"provider": "openrouter", "model": "deepseek/deepseek-r1:free"}, # DeepSeek R1(免费,高级推理能力)
{"provider": "openrouter", "model": "meta-llama/llama-3.3-70b-instruct:free"} # Meta Llama 3.3 70B(免费)
],
consensus_threshold=0.7,
max_discussion_rounds=2
)
# 从多模型协商中获取最终的共识细胞类型注释
final_annotations = consensus_results["consensus"]
# 将共识细胞类型注释整合到原始 AnnData 对象中
adata.obs['consensus_cell_type'] = adata.obs['leiden'].astype(str).map(final_annotations)
# 添加不确定性量化指标以评估注释置信度
adata.obs['consensus_proportion'] = adata.obs['leiden'].astype(str).map(consensus_results["consensus_proportion"]) # 一致性水平
adata.obs['entropy'] = adata.obs['leiden'].astype(str).map(consensus_results["entropy"]) # 注释不确定性
# 准备可视化:如果尚未计算 UMAP 嵌入,则进行计算
# UMAP 提供细胞群体的二维表示,便于可视化
if 'X_umap' not in adata.obsm:
print("正在计算 UMAP 坐标...")
# 确保先计算好近邻信息
if 'neighbors' not in adata.uns:
sc.pp.neighbors(adata, n_neighbors=10, n_pcs=30)
sc.tl.umap(adata)
print("UMAP 坐标已计算完成")
# 使用增强的美学效果可视化结果
# 基本可视化
sc.pl.umap(adata, color='consensus_cell_type', legend_loc='right', frameon=True, title='mLLMCelltype 共识注释')
# 更加定制化的可视化
import matplotlib.pyplot as plt
# 设置图形大小和样式
plt.rcParams['figure.figsize'] = (10, 8)
plt.rcParams['font.size'] = 12
# 创建更适合发表的 UMAP 图
fig, ax = plt.subplots(1, 1, figsize=(12, 10))
sc.pl.umap(adata, color='consensus_cell_type', legend_loc='on data',
frameon=True, title='mLLMCelltype 共识注释',
palette='tab20', size=50, legend_fontsize=12,
legend_fontoutline=2, ax=ax)
# 可视化不确定性指标
fig, (ax1, ax2) = plt.subplots(1, 2, figsize=(16, 7))
sc.pl.umap(adata, color='consensus_proportion', ax=ax1, title='共识比例',
cmap='viridis', vmin=0, vmax=1, size=30)
sc.pl.umap(adata, color='entropy', ax=ax2, title='注释不确定性(香农熵)',
cmap='magma', vmin=0, size=30)
plt.tight_layout()
使用单个免费 OpenRouter 模型
对于希望采用更简单方法、仅使用一个模型的用户,可以通过 OpenRouter 使用 DeepSeek R1 免费模型,而无需 API 积分:
import os
from mllmcelltype import annotate_clusters
# 注意:日志记录已自动配置
# 设置您的 OpenRouter API 密钥
os.environ["OPENROUTER_API_KEY"] = "your-openrouter-api-key"
# 定义每个聚类的标记基因
marker_genes = {
"0": ["CD3D", "CD3E", "CD3G", "CD2", "IL7R", "TCF7"], # T 细胞
"1": ["CD19", "MS4A1", "CD79A", "CD79B", "HLA-DRA", "CD74"], # B 细胞
"2": ["CD14", "LYZ", "CSF1R", "ITGAM", "CD68", "FCGR3A"] # 单核细胞
}
# 使用 DeepSeek R1 免费模型进行注释
annotations = annotate_clusters(
marker_genes=marker_genes,
species='human',
tissue='外周血',
provider='openrouter',
model='deepseek/deepseek-r1:free' # 免费且具备高级推理能力的模型
)
# 打印注释结果
for cluster, annotation in annotations.items():
print(f"Cluster {cluster}: {annotation}")
此方法使用免费模型,无需任何 API 积分。
从 AnnData 对象中提取标记基因
如果您正在使用 Scanpy 和 AnnData 对象,可以直接从 rank_genes_groups 结果中轻松提取标记基因:
import os
import scanpy as sc
from mllmcelltype import annotate_clusters
# 注意:日志记录已自动配置
# 设置您的 OpenRouter API 密钥
os.environ["OPENROUTER_API_KEY"] = "your-openrouter-api-key"
# 加载并预处理数据
adata = sc.read_h5ad('your_data.h5ad')
# 如果尚未完成预处理和聚类,可执行以下步骤:
# sc.pp.normalize_total(adata, target_sum=1e4)
# sc.pp.log1p(adata)
# sc.pp.highly_variable_genes(adata)
# sc.pp.pca(adata)
# sc.pp.neighbors(adata)
# sc.tl.leiden(adata)
# 为每个聚类寻找标记基因
sc.tl.rank_genes_groups(adata, 'leiden', method='wilcoxon')
# 提取每个聚类的前 10 个标记基因
marker_genes = {
cluster: adata.uns['rank_genes_groups']['names'][cluster][:10].tolist()
for cluster in adata.obs['leiden'].cat.categories
}
# 使用 DeepSeek R1 免费模型进行注释
annotations = annotate_clusters(
marker_genes=marker_genes,
species='human',
tissue='外周血', // 根据您的组织类型调整
provider='openrouter',
model='deepseek/deepseek-r1:free' // 免费模型
)
# 将注释添加到 AnnData 对象中
adata.obs['cell_type'] = adata.obs['leiden'].astype(str).map(annotations)
# 可视化结果
sc.pl.umap(adata, color='cell_type', legend_loc='on data',
frameon=True, title='由 DeepSeek R1 注释的细胞类型')
这种方法会自动从 rank_genes_groups 结果中提取每个聚类的前若干个差异表达基因,从而轻松地将 mLLMCelltype 集成到您的 Scanpy 工作流中。
R
注意:有关更详细的 R 教程和文档,请访问 mLLMCelltype 文档网站。
使用 Seurat 对象
# 加载所需包
library(mLLMCelltype)
library(Seurat)
library(dplyr)
library(ggplot2)
library(cowplot) # 用于 plot_grid
# 加载您预处理好的 Seurat 对象
pbmc <- readRDS("your_seurat_object.rds")
# 如果从原始数据开始,需执行预处理步骤
# pbmc <- NormalizeData(pbmc)
# pbmc <- FindVariableFeatures(pbmc, selection.method = "vst", nfeatures = 2000)
# pbmc <- ScaleData(pbmc)
# pbmc <- RunPCA(pbmc)
# pbmc <- FindNeighbors(pbmc, dims = 1:10)
# pbmc <- FindClusters(pbmc, resolution = 0.5)
# pbmc <- RunUMAP(pbmc, dims = 1:10)
# 为每个聚类寻找标记基因
pbmc_markers <- FindAllMarkers(pbmc,
only.pos = TRUE,
min.pct = 0.25,
logfc.threshold = 0.25)
# 设置缓存目录以加快处理速度
cache_dir <- "./mllmcelltype_cache"
dir.create(cache_dir, showWarnings = FALSE, recursive = TRUE)
# 选择任意支持提供商的模型
# 支持的模型包括:
# - OpenAI: 'gpt-5.2', 'gpt-5', 'gpt-4.1', 'o3-pro', 'o3', 'o4-mini', 'o1', 'o1-pro'
# - Anthropic: 'claude-opus-4-6-20260205', 'claude-sonnet-4-5-20250929', 'claude-haiku-4-5-20251001'
# - DeepSeek: 'deepseek-chat', 'deepseek-reasoner'
# - Google: 'gemini-3-pro', 'gemini-3-flash', 'gemini-2.5-pro', 'gemini-2.0-flash'
# - Qwen: 'qwen3-max', 'qwen-max-2025-01-25'
# - Stepfun: 'step-3', 'step-2-16k', 'step-2-mini'
# - Zhipu: 'glm-4.7', 'glm-4-plus'
# - MiniMax: 'minimax-m2.1', 'minimax-m2'
# - Grok: 'grok-4', 'grok-4.1', 'grok-4-heavy', 'grok-3', 'grok-3-fast', 'grok-3-mini'
# - OpenRouter: 通过单一 API 访问多家提供商的模型。格式:'provider/model-name'
# - OpenAI 模型:'openai/gpt-5.2', 'openai/gpt-5', 'openai/o3-pro', 'openai/o4-mini'
# - Anthropic 模型:'anthropic/claude-opus-4.5', 'anthropic/claude-sonnet-4.5', 'anthropic/claude-haiku-4.5'
# - Meta 模型:'meta-llama/llama-4-maverick', 'meta-llama/llama-4-scout', 'meta-llama/llama-3.3-70b-instruct'
# - Google 模型:'google/gemini-3-pro', 'google/gemini-3-flash', 'google/gemini-2.5-pro'
# - Mistral 模型:'mistralai/mistral-large', 'mistralai/magistral-medium-2506'
# - 其他模型:'deepseek/deepseek-r1', 'deepseek/deepseek-chat-v3.1', 'microsoft/mai-ds-r1'
# 使用多个 LLM 模型运行 LLMCelltype 注释
consensus_results <- interactive_consensus_annotation(
input = pbmc_markers,
tissue_name = "human PBMC", # 提供组织背景
models = c(
"claude-sonnet-4-5-20250929", # Anthropic
"gpt-5.2", # OpenAI
"gemini-3-pro", # Google
"qwen3-max" # Alibaba
),
api_keys = list(
anthropic = "your-anthropic-key",
openai = "your-openai-key",
gemini = "your-google-key",
qwen = "your-qwen-key"
),
top_gene_count = 10,
controversy_threshold = 1.0,
entropy_threshold = 1.0,
cache_dir = cache_dir
)
# 打印结果结构以了解数据
print("consensus_results 中可用字段:")
print(names(consensus_results))
# 将注释添加到 Seurat 对象
# 从 consensus_results$final_annotations 获取细胞类型注释
cluster_to_celltype_map <- consensus_results$final_annotations
# 创建新的细胞类型标识列
cell_types <- as.character(Idents(pbmc))
for (cluster_id in names(cluster_to_celltype_map)) {
cell_types[cell_types == cluster_id] <- cluster_to_celltype_map[[cluster_id]]
}
# 将细胞类型注释添加到 Seurat 对象
pbmc$cell_type <- cell_types
# 添加不确定性指标
# 提取包含指标的详细共识结果
consensus_details <- consensus_results$initial_results$consensus_results
# 为每个簇创建一个包含指标的数据框
uncertainty_metrics <- data.frame(
cluster_id = names(consensus_details),
consensus_proportion = sapply(consensus_details, function(res) res$consensus_proportion),
entropy = sapply(consensus_details, function(res) res$entropy)
)
# 为每个细胞添加不确定性指标
# 注意:seurat_clusters 是 FindClusters() 函数自动创建的元数据列
# 它包含在聚类过程中分配给每个细胞的簇 ID
# 我们在这里使用它将簇级别的指标(consensus_proportion 和 entropy)映射到单个细胞上
# 如果没有 seurat_clusters 列(例如,您使用了不同的聚类方法),
# 您可以使用活动身份(Idents)或其他元数据中的簇分配:
# 选项 1:使用活动身份
# current_clusters <- as.character(Idents(pbmc))
# 选项 2:使用包含簇 ID 的其他元数据列
# current_clusters <- pbmc$your_cluster_column
# 对于本示例,我们使用标准的 seurat_clusters 列:
current_clusters <- pbmc$seurat_clusters # 获取每个细胞的簇 ID
# 将每个细胞的簇 ID 与 uncertainty_metrics 中对应的指标匹配
pbmc$consensus_proportion <- uncertainty_metrics$consensus_proportion[match(current_clusters, uncertainty_metrics$cluster_id)]
pbmc$entropy <- uncertainty_metrics$entropy[match(current_clusters, uncertainty_metrics$cluster_id)]
# 保存结果以备将来使用
saveRDS(consensus_results, "pbmc_mLLMCelltype_results.rds")
saveRDS(pbmc, "pbmc_annotated.rds")
# 使用 SCpubr 可视化结果,生成适合发表的图表
if (!requireNamespace("SCpubr", quietly = TRUE)) {
remotes::install_github("enblacar/SCpubr")
}
library(SCpubr)
library(viridis) # 用于颜色方案
# 基本 UMAP 可视化,采用默认设置
pdf("pbmc_basic_annotations.pdf", width=8, height=6)
SCpubr::do_DimPlot(sample = pbmc,
group.by = "cell_type",
label = TRUE,
legend.position = "right") +
ggtitle("mLLMCelltype Consensus Annotations")
dev.off()
# 更加定制化的可视化,增强样式效果
pdf("pbmc_custom_annotations.pdf", width=8, height=6)
SCpubr::do_DimPlot(sample = pbmc,
group.by = "cell_type",
label = TRUE,
label.box = TRUE,
legend.position = "right",
pt.size = 1.0,
border.size = 1,
font.size = 12) +
ggtitle("mLLMCelltype Consensus Annotations") +
theme(plot.title = element_text(hjust = 0.5))
dev.off()
# 使用增强版 SCpubr 图表可视化不确定性指标
# 获取细胞类型并创建命名颜色 palette
cell_types <- unique(pbmc$cell_type)
color_palette <- viridis::viridis(length(cell_types))
names(color_palette) <- cell_types
# 使用 SCpubr 进行细胞类型标注
p1 <- SCpubr::do_DimPlot(sample = pbmc,
group.by = "cell_type",
label = TRUE,
legend.position = "bottom", # 将图例放在底部
pt.size = 1.0,
label.size = 4, # 较小的标签字体大小
label.box = TRUE, # 为标签添加背景框以提高可读性
repel = TRUE, # 使标签相互排斥以避免重叠
colors.use = color_palette,
plot.title = "Cell Type") +
theme(plot.title = element_text(hjust = 0.5, margin = margin(b = 15, t = 10)),
legend.text = element_text(size = 8),
legend.key.size = unit(0.3, "cm"),
plot.margin = unit(c(0.8, 0.8, 0.8, 0.8), "cm"))
# 使用SCpubr绘制共识比例特征图
p2 <- SCpubr::do_FeaturePlot(sample = pbmc,
features = "consensus_proportion",
order = TRUE,
pt.size = 1.0,
enforce_symmetry = FALSE,
legend.title = "Consensus",
plot.title = "Consensus Proportion",
sequential.palette = "YlGnBu", # 黄-绿-蓝渐变,遵循Nature Methods标准
sequential.direction = 1, # 由浅到深的方向
min.cutoff = min(pbmc$consensus_proportion), # 设置最小值
max.cutoff = max(pbmc$consensus_proportion), # 设置最大值
na.value = "lightgrey") + # 缺失值的颜色
theme(plot.title = element_text(hjust = 0.5, margin = margin(b = 15, t = 10)),
plot.margin = unit(c(0.8, 0.8, 0.8, 0.8), "cm"))
# 使用SCpubr绘制香农熵特征图
p3 <- SCpubr::do_FeaturePlot(sample = pbmc,
features = "entropy",
order = TRUE,
pt.size = 1.0,
enforce_symmetry = FALSE,
legend.title = "Entropy",
plot.title = "Shannon Entropy",
sequential.palette = "OrRd", # 橙-红渐变,遵循Nature Methods标准
sequential.direction = -1, # 由深到浅的方向(反转)
min.cutoff = min(pbmc$entropy), # 设置最小值
max.cutoff = max(pbmc$entropy), # 设置最大值
na.value = "lightgrey") + # 缺失值的颜色
theme(plot.title = element_text(hjust = 0.5, margin = margin(b = 15, t = 10)),
plot.margin = unit(c(0.8, 0.8, 0.8, 0.8), "cm"))
# 将图表以相等宽度组合在一起
pdf("pbmc_uncertainty_metrics.pdf", width=18, height=7)
combined_plot <- cowplot::plot_grid(p1, p2, p3, ncol = 3, rel_widths = c(1.2, 1.2, 1.2))
print(combined_plot)
dev.off()
使用CSV输入
您也可以直接使用CSV文件与mLLMCelltype配合使用,而无需Seurat,这在您已经拥有CSV格式的标记基因时非常有用:
# 安装最新版本的mLLMCelltype
devtools::install_github("cafferychen777/mLLMCelltype", subdir = "R", force = TRUE)
# 加载必要的包
library(mLLMCelltype)
# 配置统一日志记录(可选——未指定时使用默认设置)
configure_logger(level = "INFO", console_output = TRUE, json_format = TRUE)
# 创建缓存目录
cache_dir <- "path/to/your/cache"
dir.create(cache_dir, showWarnings = FALSE, recursive = TRUE)
# 读取CSV文件内容
markers_file <- "path/to/your/markers.csv"
file_content <- readLines(markers_file)
# 跳过标题行
data_lines <- file_content[-1]
# 将数据转换为列表格式,以数字索引作为键
marker_genes_list <- list()
cluster_names <- c()
# 首先收集所有簇名
for(line in data_lines) {
parts <- strsplit(line, ",", fixed = TRUE)[[1]]
cluster_names <- c(cluster_names, parts[1])
}
# 然后创建带有数字索引的marker_genes_list
for(i in 1:length(data_lines)) {
line <- data_lines[i]
parts <- strsplit(line, ",", fixed = TRUE)[[1]]
# 第一部分是簇名
cluster_name <- parts[1]
# 使用原始簇ID作为键(保留输入ID不变)
cluster_id <- as.character(cluster_name)
# 剩余部分是基因
genes <- parts[-1]
# 过滤掉NA和空字符串
genes <- genes[!is.na(genes) & genes != ""]
# 添加到marker_genes_list
marker_genes_list[[cluster_id]] <- list(genes = genes)
}
# 设置API密钥
api_keys <- list(
gemini = "YOUR_GEMINI_API_KEY",
qwen = "YOUR_QWEN_API_KEY",
grok = "YOUR_GROK_API_KEY",
openai = "YOUR_OPENAI_API_KEY",
anthropic = "YOUR_ANTHROPIC_API_KEY"
)
# 使用付费模型进行共识注释
consensus_results <-
interactive_consensus_annotation(
input = marker_genes_list,
tissue_name = "your tissue type", # 例如,“human heart”
models = c("gemini-3-pro",
"gemini-3-flash",
"qwen3-max",
"grok-4",
"claude-sonnet-4-5-20250929",
"gpt-5.2"),
api_keys = api_keys,
controversy_threshold = 0.6,
entropy_threshold = 1.0,
max_discussion_rounds = 3,
cache_dir = cache_dir
)
# 或者,使用免费的OpenRouter模型(无需积分)
# 将OpenRouter API密钥添加到api_keys列表中
api_keys$openrouter <- "your-openrouter-api-key"
# 使用免费模型进行共识注释
free_consensus_results <-
interactive_consensus_annotation(
input = marker_genes_list,
tissue_name = "your tissue type", # 例如,“human heart”
models = c(
"meta-llama/llama-4-maverick:free", # Meta Llama 4 Maverick(免费)
"venice/uncensored:free", # Venice Uncensored(免费)
"deepseek/deepseek-r1:free", # DeepSeek R1(免费,高级推理能力)
"meta-llama/llama-3.3-70b-instruct:free" # Meta Llama 3.3 70B(免费)
),
api_keys = api_keys,
consensus_check_model = "deepseek/deepseek-r1:free", # 用于共识检查的免费模型
controversy_threshold = 0.6,
entropy_threshold = 1.0,
max_discussion_rounds = 2,
cache_dir = cache_dir
)
# 保存结果
saveRDS(consensus_results, "your_results.rds")
# 打印结果摘要
cat("\nResults summary:\n")
cat("Available fields:", paste(names(consensus_results), collapse=", "), "\n\n")
# 打印最终注释
cat("Final cell type annotations:\n")
for(cluster in names(consensus_results$final_annotations)) {
cat(sprintf("%s: %s\n", cluster, consensus_results$final_annotations[[cluster]]))
}
关于CSV格式的注意事项:
- CSV文件的第一列应包含用作索引的值(这些可以是簇名称、数字如0,1,2,3或1,2,3,4等)。
- 第一列中的值仅用于参考,并不会传递给LLM。
- 后续列应包含每个簇的标记基因。
- 包含猫心脏组织示例的CSV文件位于软件包的
inst/extdata/Cat_Heart_markers.csv中。
CSV结构示例:
cluster,gene
0,Negr1,Cask,Tshz2,Fstl1,Dse,Celf2,Hmcn2,Setbp1,Cblb
1,Palld,Grb14,Mybpc3,Ensfcag00000044939,Dcun1d2,Acacb,Slco1c1,Ppp1r3c,Sema3c,Ppp1r14c
2,Adgrf5,Tbx1,Slco2b1,Pi15,Adam23,Bmx,Pde8b,Pkhd1l1,Dtx1,Ensfcag00000051556
3,Clec2d,Trat1,Rasgrp1,Card11,Cytip,Sytl3,Tmem156,Bcl11b,Lcp1,Lcp2
您可以在R脚本中通过以下方式访问示例数据:
system.file("extdata", "Cat_Heart_markers.csv", package = "mLLMCelltype")
使用单个LLM模型
如果您只想使用单个LLM模型而不是共识方法,可以使用annotate_cell_types()函数。这在您只拥有一份API密钥或更倾向于特定模型时非常有用:
# 加载所需包
library(mLLMCelltype)
library(Seurat)
# 加载预处理好的Seurat对象
pbmc <- readRDS("your_seurat_object.rds")
# 为每个簇寻找标记基因
pbmc_markers <- FindAllMarkers(pbmc,
only.pos = TRUE,
min.pct = 0.25,
logfc.threshold = 0.25)
# 从任何支持的提供商中选择一个模型
# 支持的模型包括:
# - OpenAI: 'gpt-5.2', 'gpt-5', 'gpt-4.1', 'o3-pro', 'o3', 'o4-mini', 'o1', 'o1-pro'
# - Anthropic: 'claude-opus-4-6-20260205', 'claude-sonnet-4-5-20250929', 'claude-haiku-4-5-20251001'
# - DeepSeek: 'deepseek-chat', 'deepseek-reasoner'
# - Google: 'gemini-3-pro', 'gemini-3-flash', 'gemini-2.5-pro', 'gemini-2.0-flash'
# - Qwen: 'qwen3-max', 'qwen-max-2025-01-25'
# - Stepfun: 'step-3', 'step-2-16k', 'step-2-mini'
# - Zhipu: 'glm-4.7', 'glm-4-plus'
# - MiniMax: 'minimax-m2.1', 'minimax-m2'
# - Grok: 'grok-4', 'grok-4.1', 'grok-4-heavy', 'grok-3', 'grok-3-fast', 'grok-3-mini'
# - OpenRouter: 通过单一API访问多个提供商的模型。格式为:'provider/model-name'
# - OpenAI模型:'openai/gpt-5.2', 'openai/gpt-5', 'openai/o3-pro', 'openai/o4-mini'
# - Anthropic模型:'anthropic/claude-opus-4.5', 'anthropic/claude-sonnet-4.5', 'anthropic/claude-haiku-4.5'
# - Meta模型:'meta-llama/llama-4-maverick', 'meta-llama/llama-4-scout', 'meta-llama/llama-3.3-70b-instruct'
# - Google模型:'google/gemini-3-pro', 'google/gemini-3-flash', 'google/gemini-2.5-pro'
# - Mistral模型:'mistralai/mistral-large', 'mistralai/magistral-medium-2506'
# - 其他模型:'deepseek/deepseek-r1', 'deepseek/deepseek-chat-v3.1', 'microsoft/mai-ds-r1'
# 使用单个LLM模型运行细胞类型注释
single_model_results <- annotate_cell_types(
input = pbmc_markers,
tissue_name = "human PBMC", # 提供组织背景
model = "claude-sonnet-4-5-20250929", # 指定单个模型(Claude Sonnet 4.5)
api_key = "your-anthropic-key", # 直接提供API密钥
top_gene_count = 10
)
# 使用免费的OpenRouter模型
free_model_results <- annotate_cell_types(
input = pbmc_markers,
tissue_name = "human PBMC",
model = "meta-llama/llama-4-maverick:free", # 带有:free后缀的免费模型
api_key = "your-openrouter-key",
top_gene_count = 10
)
# 打印结果
print(single_model_results)
# 将注释添加到Seurat对象
# single_model_results是一个字符向量,每个簇对应一个注释
pbmc$cell_type <- plyr::mapvalues(
x = as.character(Idents(pbmc)),
from = names(single_model_results),
to = single_model_results
)
# 可视化结果
DimPlot(pbmc, group.by = "cell_type", label = TRUE) +
ggtitle("由单个LLM模型注释的细胞类型")
比较不同模型
你还可以通过多次运行annotate_cell_types()并使用不同的模型来比较注释结果:
# 定义要测试的模型
models_to_test <- c(
"claude-sonnet-4-5-20250929", # Anthropic
"gpt-5.2", # OpenAI
"gemini-3-pro", # Google
"qwen3-max" # Alibaba
)
# 不同提供商的API密钥
api_keys <- list(
anthropic = "your-anthropic-key",
openai = "your-openai-key",
gemini = "your-gemini-key",
qwen = "your-qwen-key"
)
# 测试每个模型并存储结果
results <- list()
for (model in models_to_test) {
provider <- get_provider(model)
api_key <- api_keys[[provider]]
# 运行注释
results[[model]] <- annotate_cell_types(
input = pbmc_markers,
tissue_name = "human PBMC",
model = model,
api_key = api_key,
top_gene_count = 10
)
# 添加到Seurat对象
column_name <- paste0("cell_type_", gsub("[^a-zA-Z0-9]", "_", model))
pbmc[[column_name]] <- plyr::mapvalues(
x = as.character(Idents(pbmc)),
from = names(results[[model]]),
to = results[[model]]
)
}
高级共识配置:指定共识检查模型
consensus_check_model参数(R)/ consensus_model参数(Python)允许你指定用于共识检查和讨论调解的LLM模型。此参数对共识注释的准确性非常重要,因为共识检查模型:
- 评估不同细胞类型注释之间的语义相似性
- 计算共识指标(比例和熵)
- 调解并综合各模型对争议簇的讨论
- 在模型意见不一致时做出最终决定
我们建议使用功能强大的模型进行共识检查,因为这会直接影响注释质量。
推荐用于共识检查的模型
- Anthropic:
claude-opus-4-6-20260205,claude-sonnet-4-5-20250929 - OpenAI:
o1,o1-pro,gpt-5.2,gpt-4.1 - Google:
gemini-3-pro,gemini-3-flash - 其他:
deepseek-r1/deepseek-reasoner,qwen3-max,grok-4
R包使用示例
# 示例1:指定共识检查模型
consensus_results <- interactive_consensus_annotation(
input = marker_genes_list,
tissue_name = "human brain",
models = c("gpt-5.2", "claude-sonnet-4-5-20250929", "gemini-3-pro", "qwen3-max"),
api_keys = api_keys,
consensus_check_model = "claude-sonnet-4-5-20250929",
controversy_threshold = 0.7,
entropy_threshold = 1.0
)
# 示例2:使用替代的共识检查模型
consensus_results <- interactive_consensus_annotation(
input = marker_genes_list,
tissue_name = "mouse liver",
models = c("gpt-5.2", "gemini-3-pro", "qwen3-max"),
api_keys = api_keys,
consensus_check_model = "claude-sonnet-4-5-20250929",
controversy_threshold = 0.7,
entropy_threshold = 1.0
)
# 示例3:使用OpenAI的推理模型
consensus_results <- interactive_consensus_annotation(
input = marker_genes_list,
tissue_name = "human免疫细胞",
models = c("gpt-5.2", "claude-sonnet-4-5-20250929", "gemini-3-pro"),
api_keys = api_keys,
consensus_check_model = "o1",
controversy_threshold = 0.7,
entropy_threshold = 1.0
)
Python包使用示例
# 示例1:指定共识模型
consensus_results = interactive_consensus_annotation(
marker_genes=marker_genes,
species="human",
tissue="brain",
models=["gpt-5.2", "claude-sonnet-4-5-20250929", "gemini-3-pro", "qwen3-max"],
consensus_model="claude-sonnet-4-5-20250929",
consensus_threshold=0.7,
entropy_threshold=1.0
)
# 示例2:使用字典格式
consensus_results = interactive_consensus_annotation(
marker_genes=marker_genes,
species="mouse",
tissue="肝脏",
models=["gpt-5.2", "gemini-3-pro", "qwen3-max"],
consensus_model={"provider": "anthropic", "model": "claude-sonnet-4-5-20250929"},
consensus_threshold=0.7,
entropy_threshold=1.0
)
示例 3:使用 Google 的模型进行共识判断
consensus_results = interactive_consensus_annotation( marker_genes=marker_genes, species="human", tissue="heart", models=["gpt-5.2", "claude-sonnet-4-5-20250929", "qwen3-max"], consensus_model={"provider": "google", "model": "gemini-3-pro"}, consensus_threshold=0.7, entropy_threshold=1.0 )
示例 4:默认行为(使用 Qwen 并回退)
consensus_results = interactive_consensus_annotation( marker_genes=marker_genes, species="human", tissue="blood", models=["gpt-5.2", "claude-sonnet-4-5-20250929", "gemini-3-pro"], # 如果未指定,则默认使用 qwen3-max,并以 claude-sonnet-4-5-20250929 作为回退 consensus_threshold=0.7, entropy_threshold=1.0 )
#### 关于共识模型选择的注意事项
1. **模型可用性**:请确保您拥有所选共识模型的 API 访问权限。如果首选模型不可用,系统将使用回退模型。
2. **一致性**:在一个项目中,所有共识检查应使用相同的模型,以保证评估标准的一致性。
3. **默认行为**:
- R:若未指定,默认使用 `models` 列表中的第一个模型。
- Python:默认使用 `qwen3-max`,并以 `claude-sonnet-4-5-20250929` 作为回退。
共识检查模型必须能够准确评估不同细胞类型名称之间的语义相似性(例如,识别出“T 淋巴细胞”和“T 细胞”指代的是同一种细胞类型),理解生物学背景,并综合多个模型的讨论结果。
### 高级功能:簇选择与缓存控制(v1.3.1)
mLLMCelltype v1.3.1 引入了两个参数,使您能够对注释过程进行精细控制:
#### 1. **clusters_to_analyze** - 选择性簇分析
此参数允许您精确指定要分析的簇,而无需手动过滤输入数据:
```r
# 示例:专注于特定簇进行 T 细胞亚型分类
consensus_results <- interactive_consensus_annotation(
input = pbmc_markers,
tissue_name = "human PBMC - T cell subtypes",
models = c("gpt-5.2", "claude-sonnet-4-5-20250929"),
api_keys = api_keys,
clusters_to_analyze = c(0, 1, 7), # 仅分析 T 细胞簇
controversy_threshold = 0.7
)
# 示例:用不同上下文重新分析有争议的簇
consensus_results <- interactive_consensus_annotation(
input = pbmc_markers,
tissue_name = "activated immune cells",
models = c("gpt-5.2", "claude-sonnet-4-5-20250929", "gemini-3-pro"),
api_keys = api_keys,
clusters_to_analyze = c("3", "5"), # 专注于特定簇
cache_dir = "consensus_cache"
)
优点:
- 无需手动对数据进行子集化
- 保持原始簇编号不变
- 仅分析相关簇,从而减少 API 调用次数和成本
- 适用于对特定细胞群体进行迭代优化
2. force_rerun - 强制绕过缓存进行全新分析
此参数会强制重新分析有争议的簇,忽略缓存结果:
# 示例:初始的广泛分析
initial_results <- interactive_consensus_annotation(
input = markers,
tissue_name = "human brain",
models = c("gpt-5.2", "claude-sonnet-4-5-20250929"),
api_keys = api_keys,
use_cache = TRUE
)
# 示例:结合特定亚型上下文重新分析
subtype_results <- interactive_consensus_annotation(
input = markers,
tissue_name = "human brain - neuronal subtypes",
models = c("gpt-5.2", "claude-sonnet-4-5-20250929"),
api_keys = api_keys,
clusters_to_analyze = c(2, 3, 5), # 神经元簇
force_rerun = TRUE, # 强制全新分析,尽管存在缓存
use_cache = TRUE # 对于非争议簇仍可利用缓存以提升性能
)
重要提示:
force_rerun仅影响需要 LLM 讨论的争议簇- 非争议簇仍将使用缓存以提高效率
- 在更改组织上下文或专注于亚型时非常有用
- 可与
clusters_to_analyze结合使用,实现有针对性的重新分析
常见应用场景
- 迭代式亚型分类工作流:
# 步骤 1:通用细胞类型注释
general_types <- interactive_consensus_annotation(
input = data,
tissue_name = "human PBMC",
models = models,
api_keys = api_keys
)
# 步骤 2:聚焦 T 细胞及其亚型上下文
t_cell_subtypes <- interactive_consensus_annotation(
input = data,
tissue_name = "human T lymphocytes",
models = models,
api_keys = api_keys,
clusters_to_analyze = c(0, 1, 4, 7), # 来自步骤 1 的 T 细胞簇
force_rerun = TRUE # 结合 T 细胞上下文进行全新分析
)
# 步骤 3:进一步细化 CD8+ T 细胞亚型
cd8_subtypes <- interactive_consensus_annotation(
input = data,
tissue_name = "human CD8+ T cells - activation states",
models = models,
api_keys = api_keys,
clusters_to_analyze = c(1, 4), # CD8+ 簇
force_rerun = TRUE
)
- 经济高效的重新分析:
# 仅重新分析那些存在争议的簇
controversial <- initial_results$controversial_clusters
refined_results <- interactive_consensus_annotation(
input = data,
tissue_name = "human PBMC - refined",
models = c("gpt-5.2", "claude-sonnet-4-5-20250929", "gemini-3-pro"),
api_keys = api_keys,
clusters_to_analyze = controversial, # 仅针对有争议的簇
force_rerun = TRUE,
consensus_check_model = "claude-sonnet-4-5-20250929"
)
可视化示例
细胞类型注释可视化
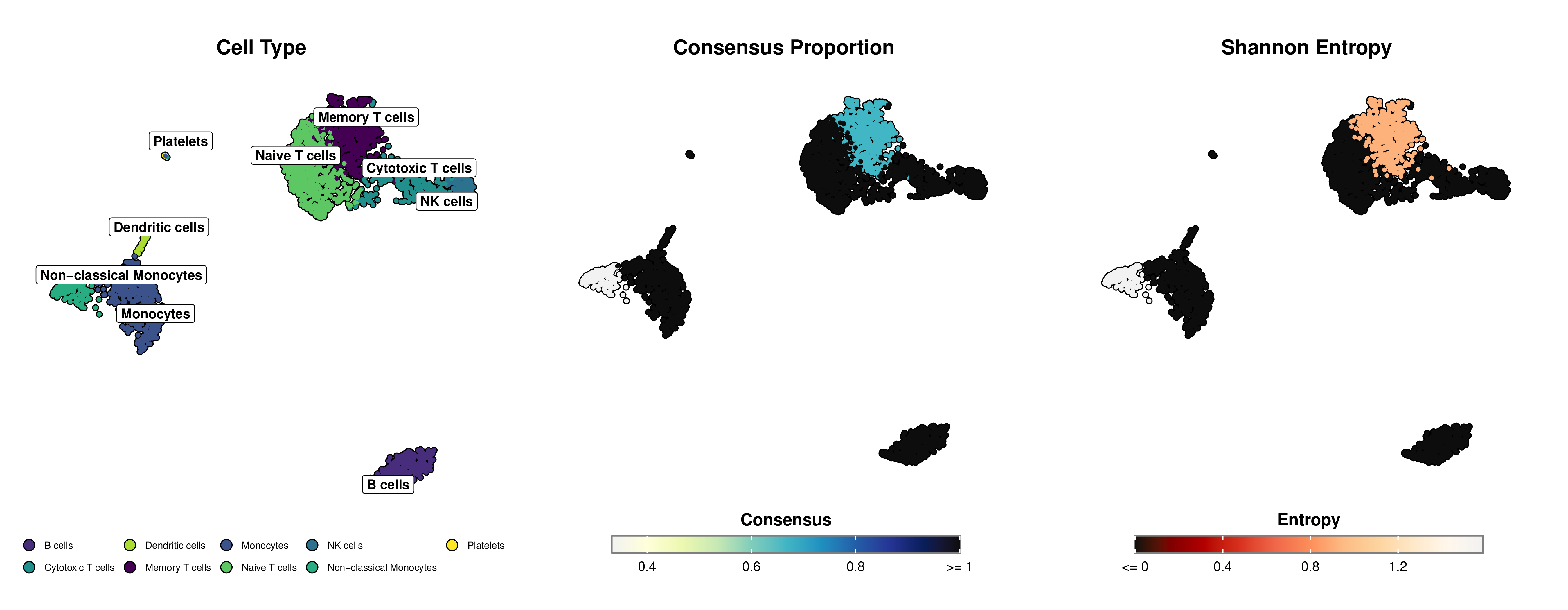
以下是使用 mLLMCelltype 和 SCpubr 创建的可用于发表的可视化示例,展示了细胞类型注释以及不确定性指标(共识比例和香农熵):
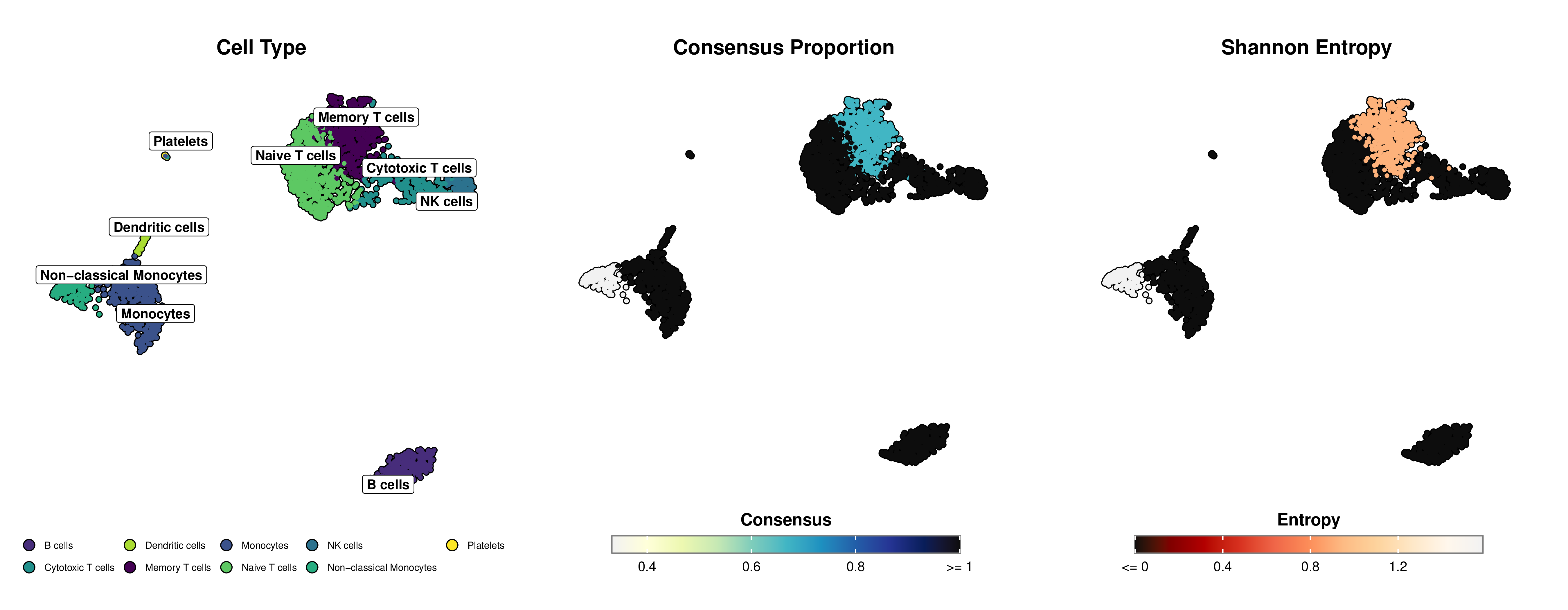
图:左侧面板显示 UMAP 投影上的细胞类型注释。中间面板使用黄绿蓝渐变色表示共识比例(蓝色越深表示 LLM 之间的一致性越高)。右侧面板使用橙红色渐变色表示香农熵(红色越深表示不确定性越低,橙色越浅表示不确定性越高)。
标记基因可视化
mLLMCelltype 包含与共识注释流程集成的标记基因可视化函数:
# 加载所需库
library(mLLMCelltype)
library(Seurat)
library(ggplot2)
# 运行共识注释后
consensus_results <- interactive_consensus_annotation(
input = markers_df,
tissue_name = "human PBMC",
models = c("anthropic/claude-sonnet-4.5", "openai/gpt-5.2"),
api_keys = list(openrouter = "your_api_key")
)
# 使用 Seurat 创建标记基因可视化
# 将共识注释添加到 Seurat 对象中
cluster_ids <- as.character(Idents(pbmc_data))
cell_type_annotations <- consensus_results$final_annotations[cluster_ids]
# 处理缺失的注释
if (any(is.na(cell_type_annotations))) {
na_mask <- is.na(cell_type_annotations)
cell_type_annotations[na_mask] <- paste("Cluster", cluster_ids[na_mask])
}
# 添加到 Seurat 对象
pbmc_data@meta.data$cell_type_consensus <- cell_type_annotations
# 创建标记基因点图
DotPlot(pbmc_data,
features = top_markers,
group.by = "cell_type_consensus") +
RotatedAxis()
# 创建标记基因热图
DoHeatmap(pbmc_data,
features = top_markers,
group.by = "cell_type_consensus")
标记基因可视化功能:
- 点图(DotPlot):同时展示每个基因在细胞中表达的百分比(点的大小)和平均表达水平(颜色强度)
- 热图(Heatmap):显示归一化后的表达值,并对基因和细胞类型进行聚类
- 集成性:可直接与添加到 Seurat 对象中的共识注释结果配合使用
- 标准 Seurat 函数:采用熟悉的 Seurat 可视化函数,保持一致性
有关详细说明和高级自定义选项,请参阅 可视化指南。
引用
如果您在研究中使用 mLLMCelltype,请引用以下内容:
@article{Yang2025.04.10.647852,
author = {Yang, Chen and Zhang, Xianyang and Chen, Jun},
title = {大型语言模型共识显著提升单细胞 RNA 测序数据的细胞类型注释准确度},
elocation-id = {2025.04.10.647852},
year = {2025},
doi = {10.1101/2025.04.10.647852},
publisher = {冷泉港实验室},
URL = {https://www.biorxiv.org/content/early/2025/04/17/2025.04.10.647852},
journal = {bioRxiv}
}
您也可以使用以下纯文本格式引用:
Yang, C., Zhang, X., & Chen, J. (2025). 大型语言模型共识显著提升单细胞 RNA 测序数据的细胞类型注释准确度。bioRxiv。阅读我们在 bioRxiv 上的完整研究论文
贡献
我们欢迎社区成员的贡献。您可以通过多种方式参与 mLLMCelltype 的开发:
报告问题
如果您遇到任何错误、有功能需求或对 mLLMCelltype 的使用有任何疑问,请在我们的 GitHub 仓库中 提交一个问题。报告问题时,请提供以下信息:
- 清晰的问题描述
- 复现问题的步骤
- 预期行为与实际行为的对比
- 您的操作系统及软件包版本信息
- 相关的代码片段或错误信息
拉取请求
我们也鼓励您通过拉取请求贡献代码改进或新功能:
- 克隆仓库并创建分支(
git checkout -b feature/amazing-feature) - 提交更改(
git commit -m '添加一项很棒的功能') - 推送到分支(
git push origin feature/amazing-feature) - 打开拉取请求
可贡献的方向
以下是一些特别需要帮助的领域:
- 增加对新 LLM 模型的支持
- 改进文档和示例
- 优化性能
- 增加新的可视化选项
- 扩展针对特定细胞类型或组织的功能
- 将文档翻译成其他语言
代码风格
请遵循仓库中现有的代码风格。对于 R 代码,我们通常遵循 tidyverse 风格指南;对于 Python 代码,则遵循 PEP 8。
社区
加入我们的 Discord 社区,讨论 mLLMCelltype 和单细胞 RNA 测序分析相关话题。
感谢您为 mLLMCelltype 的改进贡献力量!
版本历史
v2.0.02026/02/08v1.2.92025/07/01v1.2.82025/06/24v1.2.42025/05/25v1.2.02025/04/30v1.1.42025/04/28常见问题
相似工具推荐
openclaw
OpenClaw 是一款专为个人打造的本地化 AI 助手,旨在让你在自己的设备上拥有完全可控的智能伙伴。它打破了传统 AI 助手局限于特定网页或应用的束缚,能够直接接入你日常使用的各类通讯渠道,包括微信、WhatsApp、Telegram、Discord、iMessage 等数十种平台。无论你在哪个聊天软件中发送消息,OpenClaw 都能即时响应,甚至支持在 macOS、iOS 和 Android 设备上进行语音交互,并提供实时的画布渲染功能供你操控。 这款工具主要解决了用户对数据隐私、响应速度以及“始终在线”体验的需求。通过将 AI 部署在本地,用户无需依赖云端服务即可享受快速、私密的智能辅助,真正实现了“你的数据,你做主”。其独特的技术亮点在于强大的网关架构,将控制平面与核心助手分离,确保跨平台通信的流畅性与扩展性。 OpenClaw 非常适合希望构建个性化工作流的技术爱好者、开发者,以及注重隐私保护且不愿被单一生态绑定的普通用户。只要具备基础的终端操作能力(支持 macOS、Linux 及 Windows WSL2),即可通过简单的命令行引导完成部署。如果你渴望拥有一个懂你
stable-diffusion-webui
stable-diffusion-webui 是一个基于 Gradio 构建的网页版操作界面,旨在让用户能够轻松地在本地运行和使用强大的 Stable Diffusion 图像生成模型。它解决了原始模型依赖命令行、操作门槛高且功能分散的痛点,将复杂的 AI 绘图流程整合进一个直观易用的图形化平台。 无论是希望快速上手的普通创作者、需要精细控制画面细节的设计师,还是想要深入探索模型潜力的开发者与研究人员,都能从中获益。其核心亮点在于极高的功能丰富度:不仅支持文生图、图生图、局部重绘(Inpainting)和外绘(Outpainting)等基础模式,还独创了注意力机制调整、提示词矩阵、负向提示词以及“高清修复”等高级功能。此外,它内置了 GFPGAN 和 CodeFormer 等人脸修复工具,支持多种神经网络放大算法,并允许用户通过插件系统无限扩展能力。即使是显存有限的设备,stable-diffusion-webui 也提供了相应的优化选项,让高质量的 AI 艺术创作变得触手可及。
everything-claude-code
everything-claude-code 是一套专为 AI 编程助手(如 Claude Code、Codex、Cursor 等)打造的高性能优化系统。它不仅仅是一组配置文件,而是一个经过长期实战打磨的完整框架,旨在解决 AI 代理在实际开发中面临的效率低下、记忆丢失、安全隐患及缺乏持续学习能力等核心痛点。 通过引入技能模块化、直觉增强、记忆持久化机制以及内置的安全扫描功能,everything-claude-code 能显著提升 AI 在复杂任务中的表现,帮助开发者构建更稳定、更智能的生产级 AI 代理。其独特的“研究优先”开发理念和针对 Token 消耗的优化策略,使得模型响应更快、成本更低,同时有效防御潜在的攻击向量。 这套工具特别适合软件开发者、AI 研究人员以及希望深度定制 AI 工作流的技术团队使用。无论您是在构建大型代码库,还是需要 AI 协助进行安全审计与自动化测试,everything-claude-code 都能提供强大的底层支持。作为一个曾荣获 Anthropic 黑客大奖的开源项目,它融合了多语言支持与丰富的实战钩子(hooks),让 AI 真正成长为懂上
ComfyUI
ComfyUI 是一款功能强大且高度模块化的视觉 AI 引擎,专为设计和执行复杂的 Stable Diffusion 图像生成流程而打造。它摒弃了传统的代码编写模式,采用直观的节点式流程图界面,让用户通过连接不同的功能模块即可构建个性化的生成管线。 这一设计巧妙解决了高级 AI 绘图工作流配置复杂、灵活性不足的痛点。用户无需具备编程背景,也能自由组合模型、调整参数并实时预览效果,轻松实现从基础文生图到多步骤高清修复等各类复杂任务。ComfyUI 拥有极佳的兼容性,不仅支持 Windows、macOS 和 Linux 全平台,还广泛适配 NVIDIA、AMD、Intel 及苹果 Silicon 等多种硬件架构,并率先支持 SDXL、Flux、SD3 等前沿模型。 无论是希望深入探索算法潜力的研究人员和开发者,还是追求极致创作自由度的设计师与资深 AI 绘画爱好者,ComfyUI 都能提供强大的支持。其独特的模块化架构允许社区不断扩展新功能,使其成为当前最灵活、生态最丰富的开源扩散模型工具之一,帮助用户将创意高效转化为现实。
markitdown
MarkItDown 是一款由微软 AutoGen 团队打造的轻量级 Python 工具,专为将各类文件高效转换为 Markdown 格式而设计。它支持 PDF、Word、Excel、PPT、图片(含 OCR)、音频(含语音转录)、HTML 乃至 YouTube 链接等多种格式的解析,能够精准提取文档中的标题、列表、表格和链接等关键结构信息。 在人工智能应用日益普及的今天,大语言模型(LLM)虽擅长处理文本,却难以直接读取复杂的二进制办公文档。MarkItDown 恰好解决了这一痛点,它将非结构化或半结构化的文件转化为模型“原生理解”且 Token 效率极高的 Markdown 格式,成为连接本地文件与 AI 分析 pipeline 的理想桥梁。此外,它还提供了 MCP(模型上下文协议)服务器,可无缝集成到 Claude Desktop 等 LLM 应用中。 这款工具特别适合开发者、数据科学家及 AI 研究人员使用,尤其是那些需要构建文档检索增强生成(RAG)系统、进行批量文本分析或希望让 AI 助手直接“阅读”本地文件的用户。虽然生成的内容也具备一定可读性,但其核心优势在于为机器
LLMs-from-scratch
LLMs-from-scratch 是一个基于 PyTorch 的开源教育项目,旨在引导用户从零开始一步步构建一个类似 ChatGPT 的大型语言模型(LLM)。它不仅是同名技术著作的官方代码库,更提供了一套完整的实践方案,涵盖模型开发、预训练及微调的全过程。 该项目主要解决了大模型领域“黑盒化”的学习痛点。许多开发者虽能调用现成模型,却难以深入理解其内部架构与训练机制。通过亲手编写每一行核心代码,用户能够透彻掌握 Transformer 架构、注意力机制等关键原理,从而真正理解大模型是如何“思考”的。此外,项目还包含了加载大型预训练权重进行微调的代码,帮助用户将理论知识延伸至实际应用。 LLMs-from-scratch 特别适合希望深入底层原理的 AI 开发者、研究人员以及计算机专业的学生。对于不满足于仅使用 API,而是渴望探究模型构建细节的技术人员而言,这是极佳的学习资源。其独特的技术亮点在于“循序渐进”的教学设计:将复杂的系统工程拆解为清晰的步骤,配合详细的图表与示例,让构建一个虽小但功能完备的大模型变得触手可及。无论你是想夯实理论基础,还是为未来研发更大规模的模型做准备