masif
MaSIF(分子表面相互作用指纹)是一款利用几何深度学习技术,专门用于解析蛋白质分子表面特征模式的开源工具。它主要解决了传统方法难以高效识别和预测蛋白质表面关键生物分子相互作用位点的难题,例如配体结合口袋预测、蛋白 - 蛋白相互作用位点判定以及复合物结构的超快速扫描。
该工具的核心工作流程是将复杂的蛋白质表面分解为重叠的径向补丁,为每个点赋予几何与化学特征,并通过神经网络生成独特的“指纹”描述符。这些描述符能够灵活适配不同的训练目标,从而在多种生物计算任务中实现高精度分类。相比早期版本,MaSIF 已全面采用 Python 和 TensorFlow 重构,大幅简化了安装部署流程,并提供了 PyMOL 插件和 Docker 容器支持,提升了易用性。
MaSIF 特别适合生物信息学研究人员、计算生物学开发者以及从事药物发现领域的科学家使用。对于希望探索深度学习在结构生物学中应用的研究者而言,它不仅复现了发表于《Nature Methods》的前沿成果,更提供了一个可扩展的代码框架,便于在此基础上开发新的蛋白质表面学习应用。
使用场景
某生物制药公司的结构生物学团队正致力于从海量蛋白质数据库中筛选能特异性结合新型病毒靶点的候选药物分子。
没有 masif 时
- 特征提取依赖人工:研究人员需手动定义几何或化学描述符,难以捕捉蛋白质表面复杂的非线性相互作用模式。
- 结合位点预测不准:传统算法对蛋白质 - 蛋白质相互作用(PPI)界面的识别率低,导致大量潜在活性分子被漏筛。
- 大规模筛选耗时:在数以万计的结构中进行对接模拟需要巨大的计算资源和数周时间,严重拖慢研发进度。
- 泛化能力受限:针对特定任务训练的模型难以迁移到其他结合场景,每次新目标出现都需重新设计特征工程。
使用 masif 后
- 自动解码深层模式:masif 利用几何深度学习自动将蛋白质表面分解为重叠补丁,精准编码几何与化学特征,无需人工干预。
- 高精度位点识别:通过 masif-site 模块,团队能准确预测高概率发生相互作用的表面区域,显著提升了先导化合物的命中率。
- 超快速结构扫描:借助 masif-search 的指纹匹配技术,原本需要数周的复合物构型搜索缩短至几分钟内完成,实现即时筛选。
- 架构灵活复用:同一套 masif 架构只需更换训练数据,即可无缝切换于配体预测、界面识别等不同任务,极大降低了重复开发成本。
masif 通过将蛋白质表面转化为可学习的几何指纹,彻底改变了传统基于规则的分析范式,让药物发现从“大海捞针”变为“按图索骥”。
运行环境要求
- Linux
- macOS
推荐用于训练和评估(速度提升可达 100 倍),测试环境为 NVIDIA Tesla K40,未明确具体显存和 CUDA 版本要求(依赖 TensorFlow 1.9)
最低 16GB(测试环境配置),处理完整数据集需大量磁盘空间(约 1.4TB)
快速开始
MaSIF - 分子表面相互作用指纹:基于几何深度学习解析蛋白质分子表面模式
![]()
![]()
目录:
描述
MaSIF 是一种用于解析蛋白质表面中与特定生物分子相互作用相关模式的概念验证方法。为此,MaSIF 利用了几何深度学习领域的技术。首先,MaSIF 将表面分解为具有固定测地线半径的重叠径向补丁,并为每个点分配一组几何和化学特征。随后,MaSIF 为每个表面补丁计算一个描述符,该描述符是一个编码了补丁中特征信息的向量。然后,这些描述符可以在一系列额外的层中进行处理,以对不同的相互作用进行分类。每个描述符中编码的特征以及最终输出取决于特定应用的训练数据和优化目标,这意味着相同的架构可以被重新用于各种任务。
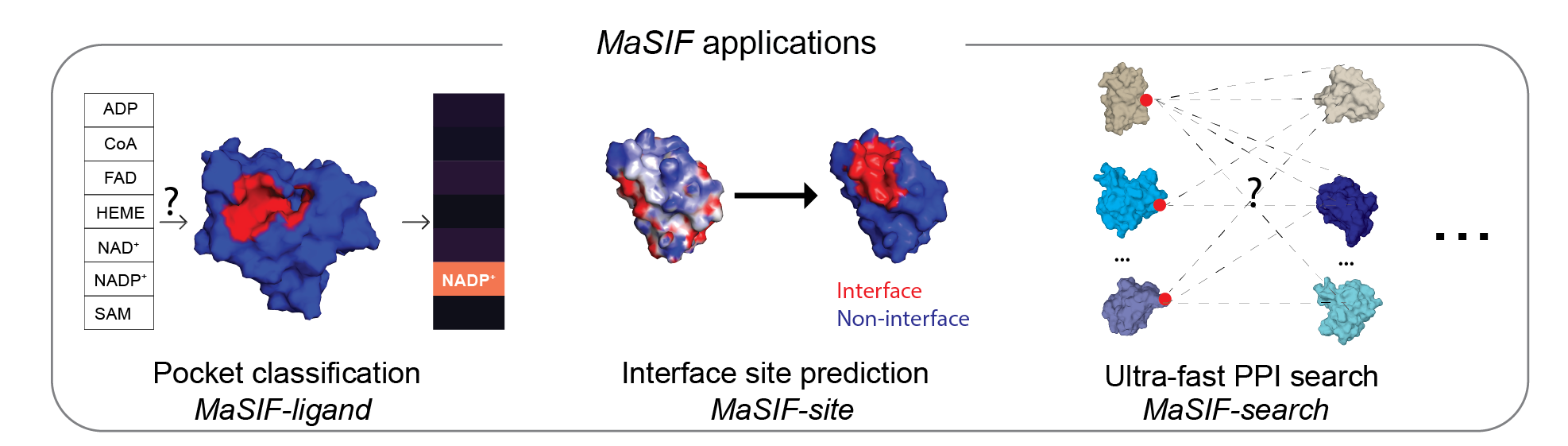
本仓库包含一个协议,用于将蛋白质结构文件转换为富含特征的表面(同时包含几何和化学特征),并将这些表面分解为补丁,以及基于 TensorFlow 的神经网络代码,以利用几何深度学习识别这些表面中的模式。为了展示该方法的潜力,我们展示了三个概念验证应用:a) 蛋白质结合口袋中的配体预测(MaSIF-ligand);b) 蛋白质-蛋白质相互作用 (PPI) 位点预测,用于预测蛋白质表面哪些补丁更有可能与其他蛋白质发生相互作用(MaSIF-site);c) 超快速表面扫描,我们使用结合伙伴的表面指纹来预测蛋白质-蛋白质复合物的结构构型(MaSIF-search)。
本仓库应能紧密复现以下研究的实验结果:
Gainza, P., Sverrisson, F., Monti, F., Rodola, E., Boscaini, D Bronstein, M. M., & Correia, B. E. (2019). 利用几何深度学习从蛋白质分子表面解密相互作用指纹。 Nat Methods 17, 184–192 (2020). https://doi.org/10.1038/s41592-019-0666-6
注:自 2020 年 2 月以来,我们通过用 Python 代码替换所有 MATLAB 代码,极大地简化了 MaSIF 的安装过程。然而,这会略微改变论文中的结果。若要完全按照已发表论文中的结果(包括预训练的神经网络)进行复现,可访问:https://github.com/pablogainza/masif_paper 。
MaSIF 采用 Apache 许可证 发布。此代码旨在作为教程,为研究人员在蛋白质表面学习任务中使用 MaSIF 提供基础。
系统及硬件要求
MaSIF 已在 Linux(Red Hat Enterprise Linux Server release 7.4,配备 Intel(R) Xeon(R) CPU E5-2650 v2 @ 2.60GHz 处理器及 16GB 内存)和 Mac OS 环境(macOS High Sierra,处理器为 2.8 GHz Intel Core i7,内存为 16GB)上进行了测试。为复现论文中的实验,所有蛋白质的数据集总共占用约 1.4 TB。
目前,MaSIF 处理每条蛋白质大约需要 2 分钟。因此,对于大型蛋白质数据集,我们建议使用分布式集群来预处理数据。一旦数据预处理完成,我们强烈建议使用 GPU 来训练或评估训练好的模型,因为其速度可能比 CPU 快达 100 倍。
软件前提条件
MaSIF 依赖于外部软件/库来处理蛋白质数据库文件和表面文件,计算化学/几何特征和坐标,以及执行神经网络计算。以下是所需库和程序的列表,以及经过测试的版本(括号内):
- Python (3.6)
- reduce (3.23)。用于为蛋白质添加质子。
- MSMS (2.6.1)。用于计算蛋白质表面。
- BioPython (1.66)。用于解析 PDB 文件。
- PyMesh (0.1.14)。用于处理 ply 格式的表面文件、属性,并对网格进行规整。
- PDB2PQR (2.1.1)、multivalue 和 APBS (1.5)。这些程序是计算静电荷所必需的。
- open3D (0.5.0.0)。主要用于 RANSAC 配准。
- Tensorflow (1.9)。用于构建、训练和评估实际的神经网络。模型是在 NVIDIA Tesla K40 GPU 上训练和评估的。
- StrBioInfo。用于解析 PDB 文件并为 MaSIF-ligand 生成生物学组装。
- Dask (2.2.0)。用于在多个线程上运行函数调用(可选,用于复现部分基准测试)。
- Pymol。此可选插件允许用户在 PyMOL 中可视化表面文件。
或者,您也可以使用 Docker 版本,这是最易于安装的方式(参见 Docker 容器)。
安装
在预先安装好依赖项后,请将以下环境变量添加到您的路径中,并根据实际情况更改目录:
export APBS_BIN=/path/to/apbs/APBS-1.5-linux64/bin/apbs
export MULTIVALUE_BIN=/path/to/apbs/APBS-1.5-linux64/share/apbs/tools/bin/multivalue
export PDB2PQR_BIN=/path/to/apbs/apbs/pdb2pqr-linux-bin64-2.1.1/pdb2pqr
export PATH=$PATH:/path/to/reduce/
export REDUCE_HET_DICT=/path/to/reduce/reduce_wwPDB_het_dict.txt
export PYMESH_PATH=/path/to/PyMesh
export MSMS_BIN=/path/to/msms/msms
export PDB2XYZRN=/path/to/msms/pdb_to_xyzrn
将 masif 克隆到本地目录:
git clone https://github.com/lpdi-epfl/masif
cd masif/
由于 MaSIF 是用 Python 编写的,因此无需编译。
方法概述
MaSIF 从蛋白质结构出发,基于溶剂可及表面(使用 MSMS 计算得到)构建一个离散化的分子表面网格,并为网格中的每个顶点分配几何和化学特征。在网格的每个顶点周围,我们提取一个测地半径为 r=9 Å 或 r=12 Å 的局部区域。随后,MaSIF 对这些局部区域应用几何深度神经网络。该神经网络由一个或多个层顺序堆叠而成;其架构中的关键组件是测地卷积,它将经典的卷积操作推广到曲面上,并以局部区域上的运算形式实现。
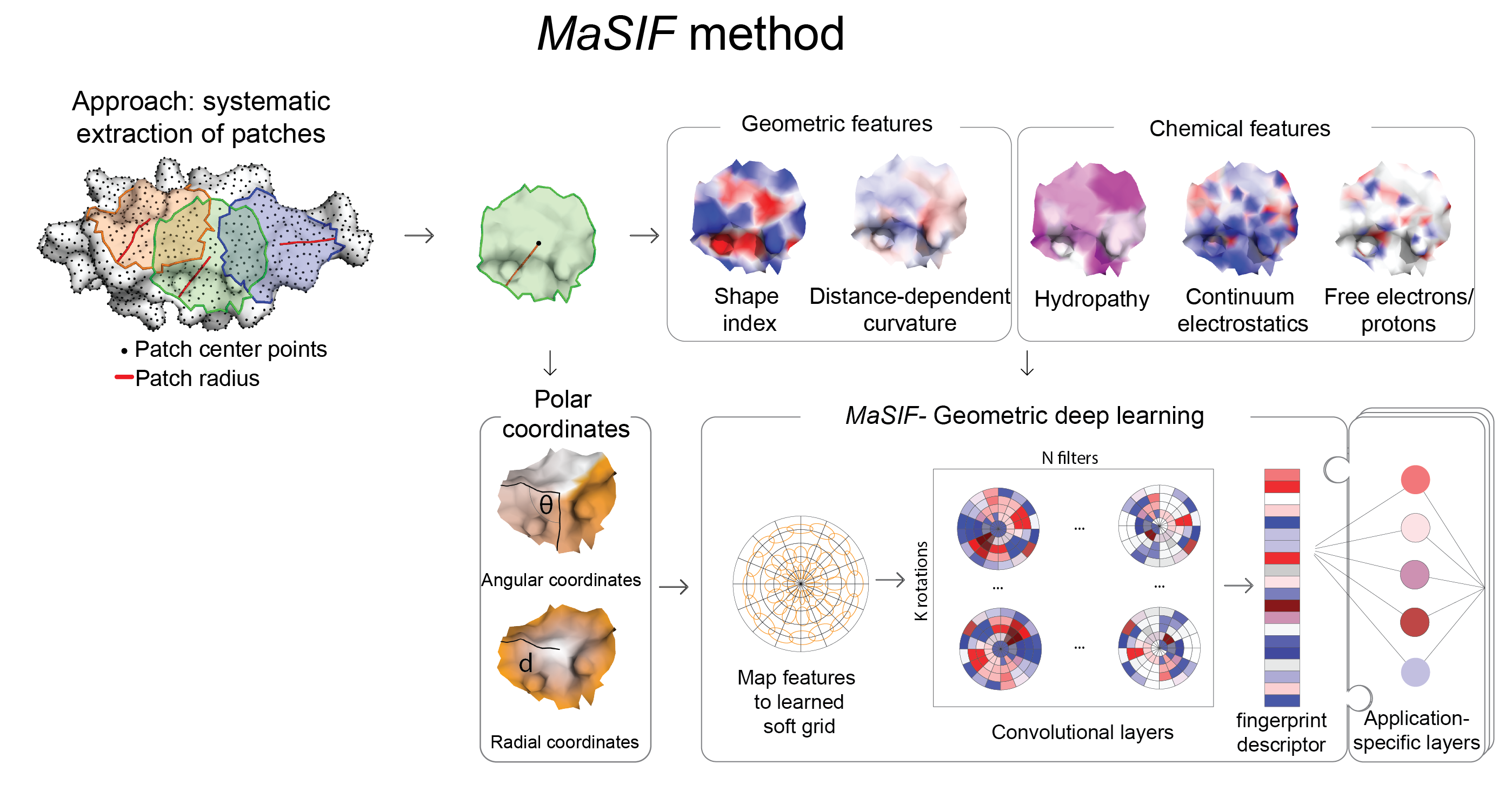
这一过程会针对不同的局部区域位置重复进行,类似于图像上的滑动窗口操作,从而在每个点生成表面指纹描述符——一个向量,用于存储中心点及其邻域的表面模式信息。模型参数通过在训练数据集上最小化特定于本文所介绍的每种应用的损失函数来确定。
MaSIF 数据准备
对于每种应用,MaSIF 需要对数据进行预处理。这包括运行一个脚本化的流程,具体步骤如下:
- 下载 PDB 文件。
- 对 PDB 进行质子化处理,提取所需的链段,利用 MSMS 对表面进行三角剖分,并计算化学特征。
- 提取每条蛋白质的所有局部区域,包括其特征和坐标。
MaSIF 的主要性能瓶颈在于上述三个步骤。其中,使用 MDS 计算角度坐标、计算泊松-玻尔兹曼静电势,以及在完成 MSMS 表面计算后对网格进行规整,是主要的性能瓶颈所在。
每个应用的数据目录(位于 masif/data/masif* 下)都包含一个用于预计算数据的脚本。
若要为单个蛋白质运行此流程(例如,PDB ID 代码为 1MBN 的 A 链),可执行以下命令:
./data_prepare_one.sh 1MBN_A_
若要对一对相互作用的蛋白质结构域运行此流程(例如,PDB ID 1AKJ 中的 A 和 B 链构成第一个结构域,D 和 E 链构成第二个结构域),则执行:
./data_prepare_one.sh 1AKJ_AB_DE
如果您可以访问集群(强烈推荐),则可以并行运行此流程。如果您的集群支持 Slurm 作业提交文件,我们会在每个应用的数据目录下提供一个 Slurm 文件,可通过 sbatch 命令运行:
sbatch data_prepare.slurm
论文中使用的大多数 PDB 文件及其对应的表面(已预先计算好化学特征)可在以下网址获取:https://doi.org/10.5281/zenodo.2625420 。未结合状态的蛋白质则可在该仓库的 data/masif_ppi_search_ub/data_preparation/00-raw_pdbs/ 目录下找到。
需要注意的是,对于大型蛋白质数据库而言,数据预处理可能会占用大量存储空间。这是因为预处理步骤会将蛋白质表面分解为重叠的局部区域,从而导致大量数据冗余。在未来的版本中,我们希望优化这一流程,实现在需要时动态进行局部区域分解,从而避免对大量磁盘空间的需求。
MaSIF 概念验证应用
MaSIF 在三个概念验证应用上进行了测试。对于每个应用,我们都提供了用于论文中主要实验的训练好的神经网络模型。
MaSIF-ligand
cd data/masif_site/
训练集和测试集中使用的 PDB ID 及链的信息以 NumPy 格式存储在以下路径:
data/masif_ligand/lists/test_pdbs_sequence.npy
data/masif_ligand/lists/train_pdbs_sequence.npy
data/masif_ligand/lists/val_pdbs_sequence.npy
这些文件均可使用 numpy.load 函数读取。
请预计算数据集(参见 MaSIF 数据准备),理想情况下使用 Slurm:
sbatch prepare_data.slurm
请确保拥有足够的磁盘空间,约 400GB。
数据预处理完成后,MaSIF-ligand 需要生成用于训练的 TensorFlow TFRecords。为此,您可以运行 Slurm 作业,或直接执行 make_tfrecord.slurm 文件中的命令:
sbatch make_tfrecord.slurm
TFRecords 预计算完成后,即可开始网络训练,强烈建议使用 GPU:
sbatch train_model.slurm
评估神经网络时,请运行:
sbatch evaluate_test.slurm
评估结果将保存在 data/masif_ligand/test_set_predictions/ 目录下,每个输入蛋白质结构对应两个 NumPy 文件,例如:
5LXM_AD_labels.npy
5LXM_AD_logits.npy
其中,labels 文件包含真实标签,logits 文件则包含预测的 logits 值。
MaSIF-site
切换到 maif-site 数据目录:
cd data/masif_site/
训练集和测试集中使用的 PDB ID 及链的信息存储在以下路径:
data/masif_site/data/lists/full_list.txt
data/masif_site/data/lists/training.txt
data/masif_site/data/lists/testing.txt
请预计算数据集(参见 MaSIF 数据准备),理想情况下使用 Slurm:
sbatch prepare_data.slurm
请确保拥有足够的磁盘空间,约 400GB。
数据预处理完成后,即可开始网络训练:
./train_nn.sh
在论文的实验中,MaSIF-site 网络训练了 40 小时。
网络训练完成后,可以对特定蛋白质进行评估。例如,要评估选定的瞬时相互作用子集,可执行:
./predict_site.sh
每个蛋白质中每个顶点的预测结果将存储在 data/masif_site/output/all_feat_3l/pred_data/ 目录下。可以根据预测结果为相应位点着色:
./color_site.sh
并将结果保存为 ply 文件,存放在 data/masif_site/output/all_feat_3l/pred_surfaces/ 目录中。
之后,您可以通过我们的 PyMOL 插件 查看这些表面。
此外,还有一个 Jupyter Notebook,其中包含代码,用于将该测试集中瞬时相互作用的预测结果与程序 SPPIDER 的结果进行比较,该 Notebook 的路径为:
masif/comparison/masif_site/masif_vs_sppider/masif_sppider_comp.ipynb
MaSIF-搜索
切换到 masif-search 数据目录。
cd data/masif_ppi_search/
训练集和测试集中使用的 PDB ID 和链的列表位于以下路径:
data/masif_ppi_searhc/data/lists/full_list.txt
data/masif_site/data/lists/training.txt
data/masif_site/data/lists/testing.txt
预计算数据集(参见 MaSIF 数据准备),理想情况下使用 Slurm 提交作业:
sbatch prepare_data.slurm
请确保有足够的磁盘空间,大约 400GB。
出于速度考虑,神经网络实际使用的数据会被缓存到一个单独的目录中。这些数据包括通过形状互补性阈值的补丁对,以及相同数量的随机补丁。该过程通过执行以下命令运行:
./cache_nn.sh nn_models.sc05.custom_params
数据缓存完成后,即可开始训练神经网络:
./train.sh nn_models.sc05.custom_params
在论文中,我们训练了约 40 小时。每当验证集的 ROC AUC 比之前保存的模型有所提高时,神经网络模型就会被保存到 nn_models/sc05/all_feat/model_data 目录下。
神经网络训练并保存完成后,可以使用以下命令为特定蛋白质计算描述符:
./compute_descriptors.sh lists/testing.txt
这些描述符将保存在 descriptors/ 目录下。
要评估第二阶段的 RANSAC 协议,请进入 masif/comparison/masif_ppi_search 目录:
cd $masif_root/comparison/masif_ppi_search/masif_descriptors/
./second_stage.sh
要复现论文中展示的大规模 PD-L1:PD1 基准测试,请执行以下操作:
cd data/masif_ppi_search/pdl1_benchmark
./run_benchmark.sh
PyMOL 插件
源代码中的 source/pymol 子目录提供了一个用于可视化蛋白质表面的 PyMOL 插件。我们在论文中展示的所有结构图都使用了该插件。此插件需要在本地计算机上安装 PyMOL。
请参阅以下教程以了解如何安装:
要加载蛋白质表面文件,请在 PyMOL 中运行以下命令:
loadply ABCD_E.ply
示例:
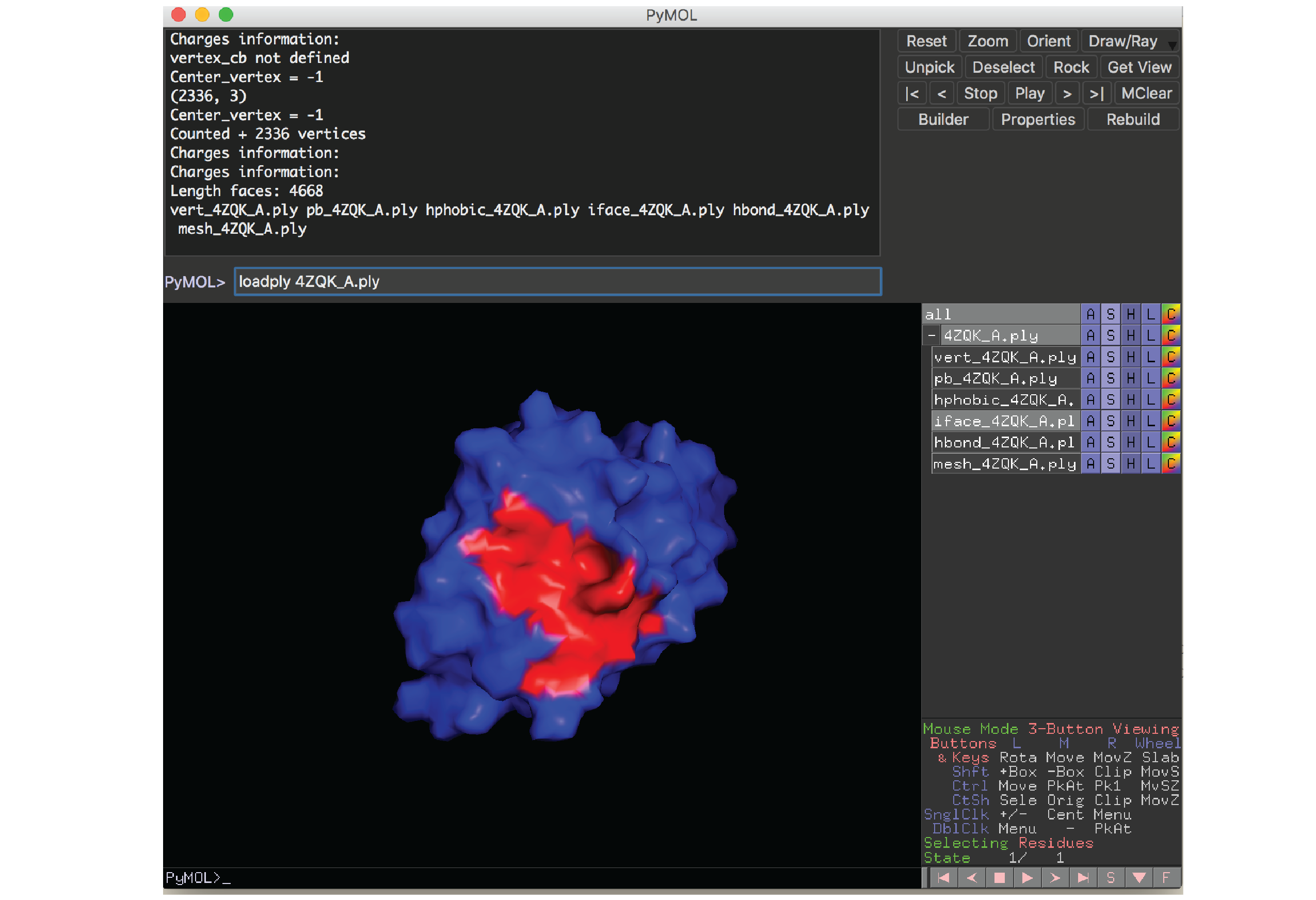
Docker 容器
测试 MaSIF 的最简单方法是通过 Docker 容器。请参阅我们的教程,了解如何复现论文结果:
许可证
MaSIF 采用 Apache v2.0 许可证 发布。
参考文献
如果您使用本代码,请引用 citation.bib 文件中的 BibTeX 条目。
版本历史
0.1.12019/10/27v0.12019/10/27常见问题
相似工具推荐
openclaw
OpenClaw 是一款专为个人打造的本地化 AI 助手,旨在让你在自己的设备上拥有完全可控的智能伙伴。它打破了传统 AI 助手局限于特定网页或应用的束缚,能够直接接入你日常使用的各类通讯渠道,包括微信、WhatsApp、Telegram、Discord、iMessage 等数十种平台。无论你在哪个聊天软件中发送消息,OpenClaw 都能即时响应,甚至支持在 macOS、iOS 和 Android 设备上进行语音交互,并提供实时的画布渲染功能供你操控。 这款工具主要解决了用户对数据隐私、响应速度以及“始终在线”体验的需求。通过将 AI 部署在本地,用户无需依赖云端服务即可享受快速、私密的智能辅助,真正实现了“你的数据,你做主”。其独特的技术亮点在于强大的网关架构,将控制平面与核心助手分离,确保跨平台通信的流畅性与扩展性。 OpenClaw 非常适合希望构建个性化工作流的技术爱好者、开发者,以及注重隐私保护且不愿被单一生态绑定的普通用户。只要具备基础的终端操作能力(支持 macOS、Linux 及 Windows WSL2),即可通过简单的命令行引导完成部署。如果你渴望拥有一个懂你
stable-diffusion-webui
stable-diffusion-webui 是一个基于 Gradio 构建的网页版操作界面,旨在让用户能够轻松地在本地运行和使用强大的 Stable Diffusion 图像生成模型。它解决了原始模型依赖命令行、操作门槛高且功能分散的痛点,将复杂的 AI 绘图流程整合进一个直观易用的图形化平台。 无论是希望快速上手的普通创作者、需要精细控制画面细节的设计师,还是想要深入探索模型潜力的开发者与研究人员,都能从中获益。其核心亮点在于极高的功能丰富度:不仅支持文生图、图生图、局部重绘(Inpainting)和外绘(Outpainting)等基础模式,还独创了注意力机制调整、提示词矩阵、负向提示词以及“高清修复”等高级功能。此外,它内置了 GFPGAN 和 CodeFormer 等人脸修复工具,支持多种神经网络放大算法,并允许用户通过插件系统无限扩展能力。即使是显存有限的设备,stable-diffusion-webui 也提供了相应的优化选项,让高质量的 AI 艺术创作变得触手可及。
everything-claude-code
everything-claude-code 是一套专为 AI 编程助手(如 Claude Code、Codex、Cursor 等)打造的高性能优化系统。它不仅仅是一组配置文件,而是一个经过长期实战打磨的完整框架,旨在解决 AI 代理在实际开发中面临的效率低下、记忆丢失、安全隐患及缺乏持续学习能力等核心痛点。 通过引入技能模块化、直觉增强、记忆持久化机制以及内置的安全扫描功能,everything-claude-code 能显著提升 AI 在复杂任务中的表现,帮助开发者构建更稳定、更智能的生产级 AI 代理。其独特的“研究优先”开发理念和针对 Token 消耗的优化策略,使得模型响应更快、成本更低,同时有效防御潜在的攻击向量。 这套工具特别适合软件开发者、AI 研究人员以及希望深度定制 AI 工作流的技术团队使用。无论您是在构建大型代码库,还是需要 AI 协助进行安全审计与自动化测试,everything-claude-code 都能提供强大的底层支持。作为一个曾荣获 Anthropic 黑客大奖的开源项目,它融合了多语言支持与丰富的实战钩子(hooks),让 AI 真正成长为懂上
ComfyUI
ComfyUI 是一款功能强大且高度模块化的视觉 AI 引擎,专为设计和执行复杂的 Stable Diffusion 图像生成流程而打造。它摒弃了传统的代码编写模式,采用直观的节点式流程图界面,让用户通过连接不同的功能模块即可构建个性化的生成管线。 这一设计巧妙解决了高级 AI 绘图工作流配置复杂、灵活性不足的痛点。用户无需具备编程背景,也能自由组合模型、调整参数并实时预览效果,轻松实现从基础文生图到多步骤高清修复等各类复杂任务。ComfyUI 拥有极佳的兼容性,不仅支持 Windows、macOS 和 Linux 全平台,还广泛适配 NVIDIA、AMD、Intel 及苹果 Silicon 等多种硬件架构,并率先支持 SDXL、Flux、SD3 等前沿模型。 无论是希望深入探索算法潜力的研究人员和开发者,还是追求极致创作自由度的设计师与资深 AI 绘画爱好者,ComfyUI 都能提供强大的支持。其独特的模块化架构允许社区不断扩展新功能,使其成为当前最灵活、生态最丰富的开源扩散模型工具之一,帮助用户将创意高效转化为现实。
gemini-cli
gemini-cli 是一款由谷歌推出的开源 AI 命令行工具,它将强大的 Gemini 大模型能力直接集成到用户的终端环境中。对于习惯在命令行工作的开发者而言,它提供了一条从输入提示词到获取模型响应的最短路径,无需切换窗口即可享受智能辅助。 这款工具主要解决了开发过程中频繁上下文切换的痛点,让用户能在熟悉的终端界面内直接完成代码理解、生成、调试以及自动化运维任务。无论是查询大型代码库、根据草图生成应用,还是执行复杂的 Git 操作,gemini-cli 都能通过自然语言指令高效处理。 它特别适合广大软件工程师、DevOps 人员及技术研究人员使用。其核心亮点包括支持高达 100 万 token 的超长上下文窗口,具备出色的逻辑推理能力;内置 Google 搜索、文件操作及 Shell 命令执行等实用工具;更独特的是,它支持 MCP(模型上下文协议),允许用户灵活扩展自定义集成,连接如图像生成等外部能力。此外,个人谷歌账号即可享受免费的额度支持,且项目基于 Apache 2.0 协议完全开源,是提升终端工作效率的理想助手。
markitdown
MarkItDown 是一款由微软 AutoGen 团队打造的轻量级 Python 工具,专为将各类文件高效转换为 Markdown 格式而设计。它支持 PDF、Word、Excel、PPT、图片(含 OCR)、音频(含语音转录)、HTML 乃至 YouTube 链接等多种格式的解析,能够精准提取文档中的标题、列表、表格和链接等关键结构信息。 在人工智能应用日益普及的今天,大语言模型(LLM)虽擅长处理文本,却难以直接读取复杂的二进制办公文档。MarkItDown 恰好解决了这一痛点,它将非结构化或半结构化的文件转化为模型“原生理解”且 Token 效率极高的 Markdown 格式,成为连接本地文件与 AI 分析 pipeline 的理想桥梁。此外,它还提供了 MCP(模型上下文协议)服务器,可无缝集成到 Claude Desktop 等 LLM 应用中。 这款工具特别适合开发者、数据科学家及 AI 研究人员使用,尤其是那些需要构建文档检索增强生成(RAG)系统、进行批量文本分析或希望让 AI 助手直接“阅读”本地文件的用户。虽然生成的内容也具备一定可读性,但其核心优势在于为机器