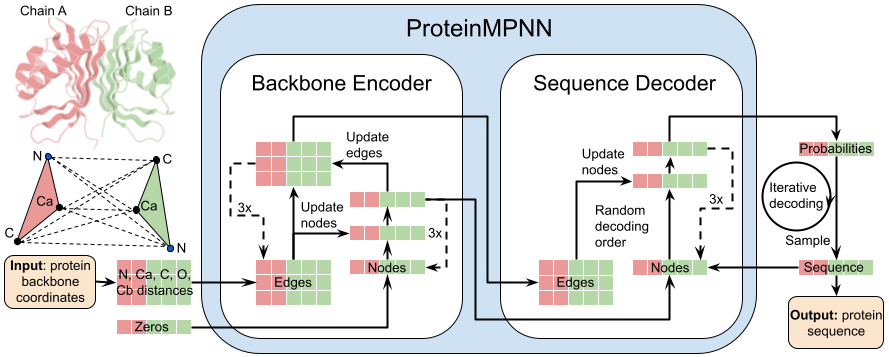
ProteinMPNN
ProteinMPNN 是一款基于深度学习的开源工具,专为蛋白质序列设计而生。它核心解决了“逆折叠”难题:在已知蛋白质三维骨架结构的前提下,精准预测出能够稳定折叠成该结构的最优氨基酸序列。这对于人工设计新型酶、药物靶点及功能蛋白至关重要。
该工具主要面向生物计算研究人员、合成生物学家及相关领域的开发者。用户只需提供蛋白质的 PDB 结构文件,即可利用预训练模型快速生成多条高成功率候选序列,同时也支持对现有序列进行稳定性评分或概率分析。
ProteinMPNN 的技术亮点在于其高效的图神经网络架构,不仅推理速度极快,且在序列恢复率等关键指标上显著优于传统方法。它提供了多种预训练权重,包括适用于完整骨架的高精度模型、仅依赖 Cα原子的简化模型,以及专门针对可溶性蛋白优化的版本。此外,项目配套了丰富的辅助脚本,支持灵活设定固定残基、添加氨基酸偏好及调整采样温度,既适合通过 Google Colab 快速上手,也便于专业用户进行本地化大规模定制开发。
使用场景
某生物制药公司的研发团队正致力于优化一种新型抗癌抗体蛋白的热稳定性,需要在保持其结合活性的前提下,重新设计部分不稳定的氨基酸序列。
没有 ProteinMPNN 时
- 试错成本极高:研究人员主要依赖经验法则或简单的能量函数进行突变预测,往往需要合成并测试数百个变体才能找到一个稳定结构,耗时数月。
- 序列多样性匮乏:传统方法生成的候选序列过于保守,难以跳出局部最优解,导致发现全新高稳定性序列的概率极低。
- 忽略骨架灵活性:现有工具难以在考虑蛋白质主链(Backbone)微小噪声的情况下生成序列,导致设计出的蛋白在实际折叠中容易失败。
- 计算资源浪费:为了获得可靠结果,团队不得不运行大量的分子动力学模拟来验证每一个手动设计的序列,算力消耗巨大。
使用 ProteinMPNN 后
- 生成效率飞跃:利用 ProteinMPNN 的
v_48_020模型,团队输入抗体 PDB 结构并固定关键结合位点,几分钟内即可生成数千个高概率的稳定序列候选者。 - 精准可控设计:通过
--conditional_probs_only参数,研究人员能精确计算特定位置的条件概率,只替换那些对稳定性贡献低且不影响功能的残基。 - 鲁棒性显著增强:开启
--backbone_noise参数引入主链噪声进行采样,使得生成的序列不仅能适应理想结构,还能容忍实际实验中的构象波动,成功率大幅提升。 - 筛选流程简化:借助
--save_score功能直接输出序列的对数概率评分,快速筛选出前 5% 的高分序列进行湿实验验证,将研发周期从数月缩短至两周。
ProteinMPNN 通过将复杂的蛋白质逆折叠问题转化为高效的概率生成任务,彻底改变了理性蛋白设计的速度与成功率。
运行环境要求
- 未说明
非绝对必需(可调整 batch_size 适应),示例命令建议安装 cudatoolkit=11.3,适用于 Titan/Quadro 等 NVIDIA GPU
未说明
快速开始
ProteinMPNN
 请阅读 ProteinMPNN 论文。
请阅读 ProteinMPNN 论文。
要运行 ProteinMPNN,请克隆此 GitHub 仓库,并安装 Python>=3.0、PyTorch 和 Numpy。
完整蛋白质骨架模型:vanilla_model_weights/v_48_002.pt, v_48_010.pt, v_48_020.pt, v_48_030.pt,soluble_model_weights/v_48_010.pt, v_48_020.pt。
仅 CA 模型:ca_model_weights/v_48_002.pt, v_48_010.pt, v_48_020.pt。启用 --ca_only 标志以使用这些模型。
辅助脚本:helper_scripts - 用于解析 PDB 文件、指定哪些链需要设计、哪些残基需要固定、添加氨基酸偏好、约束残基等的辅助函数。
代码组织:
protein_mpnn_run.py- 用于初始化和运行模型的主脚本。protein_mpnn_utils.py- 主脚本的实用函数。examples/- 简单的代码示例。inputs/- 示例输入的 PDB 文件。outputs/- 示例输出。colab_notebooks/- Google Colab 示例。training/- 用于重新训练模型的代码和数据。
protein_mpnn_run.py 的输入参数:
argparser.add_argument("--suppress_print", type=int, default=0, help="0 表示 False,1 表示 True")
argparser.add_argument("--ca_only", action="store_true", default=False, help="解析仅 CA 结构并使用仅 CA 模型(默认:False)")
argparser.add_argument("--path_to_model_weights", type=str, default="", help="模型权重文件夹路径;")
argparser.add_argument("--model_name", type=str, default="v_48_020", help="ProteinMPNN 模型名称:v_48_002、v_48_010、v_48_020、v_48_030;v_48_010 是带有 48 条边且噪声为 0.10Å 的版本")
argparser.add_argument("--use_soluble_model", action="store_true", default=False, help="标志,用于加载仅在可溶性蛋白质上训练的 ProteinMPNN 权重。")
argparser.add_argument("--seed", type=int, default=0, help="如果设置为 0,则会随机选择一个种子;")
argparser.add_argument("--save_score", type=int, default=0, help="0 表示 False,1 表示 True;将得分=-log_prob 保存为 npy 文件")
argparser.add_argument("--path_to_fasta", type=str, default="", help="以 FASTA 格式提供评分用的输入序列;例如 GGGGGG/PPPPS/WWW,分别对应按字母顺序排列并用 / 分隔的 A、B、C 链")
argparser.add_argument("--save_probs", type=int, default=0, help="0 表示 False,1 表示 True;保存 MPNN 预测的每个位置的概率")
argparser.add_argument("--score_only", type=int, default=0, help="0 表示 False,1 表示 True;仅对输入的骨架-序列对进行评分")
argparser.add_argument("--conditional_probs_only", type=int, default=0, help="0 表示 False,1 表示 True;输出条件概率 p(s_i 给定序列和骨架的其余部分)")
argparser.add_argument("--conditional_probs_only_backbone", type=int, default=0, help="0 表示 False,1 表示 True;如果为真,则输出条件概率 p(s_i 给定骨架)")
argparser.add_argument("--unconditional_probs_only", type=int, default=0, help="0 表示 False,1 表示 True;在一次前向传播中输出无条件概率 p(s_i 给定骨架)")
argparser.add_argument("--backbone_noise", type=float, default=0.00, help="添加到骨架原子上的高斯噪声的标准差")
argparser.add_argument("--num_seq_per_target", type=int, default=1, help="每个目标生成的序列数量")
argparser.add_argument("--batch_size", type=int, default=1, help="批大小;对于 Titan 或 Quadro 显卡可以设置得更高,如果显存不足则应降低该值")
argparser.add_argument("--max_length", type=int, default=200000, help="最大序列长度")
argparser.add_argument("--sampling_temp", type=str, default="0.1", help="温度字符串,如 0.2 0.25 0.5。用于氨基酸采样的温度。建议值为 0.1、0.15、0.2、0.25、0.3。较高的值会导致更多多样性。")
argparser.add_argument("--out_folder", type=str, help="输出序列的文件夹路径,例如 /home/out/")
argparser.add_argument("--pdb_path", type=str, default='', help="待设计的单个 PDB 文件路径")
argparser.add_argument("--pdb_path_chains", type=str, default='', help="定义单个 PDB 中哪些链需要设计")
argparser.add_argument("--jsonl_path", type=str, help="包含已解析为 jsonl 格式的 PDB 文件的文件夹路径")
argparser.add_argument("--chain_id_jsonl",type=str, default='', help="指定哪些链需要设计、哪些链固定不变的字典文件路径;若未指定,则所有链都将被设计。")
argparser.add_argument("--fixed_positions_jsonl", type=str, default='', help="包含固定位置的字典文件路径")
argparser.add_argument("--omit_AAs", type=list, default='X', help="指定在生成的序列中应省略哪些氨基酸,例如 'AC' 将省略丙氨酸和半胱氨酸。")
argparser.add_argument("--bias_AA_jsonl", type=str, default='', help="指定氨基酸组成偏好的字典文件路径,例如 {A: -1.1, F: 0.7} 会使 A 出现的可能性降低,而 F 出现的可能性增加。")
argparser.add_argument("--bias_by_res_jsonl", default='', help="包含按位置偏好的字典文件路径。")
argparser.add_argument("--omit_AA_jsonl", type=str, default='', help="包含在特定链索引处需要从设计中省略哪些氨基酸的字典文件路径")
argparser.add_argument("--pssm_jsonl", type=str, default='', help="包含 PSSM 的字典文件路径")
argparser.add_argument("--pssm_multi", type=float, default=0.0, help="一个介于 [0.0, 1.0] 之间的值,0.0 表示不使用 PSSM,1.0 表示忽略 MPNN 的预测")
argparser.add_argument("--pssm_threshold", type=float, default=0.0, help="一个介于负无穷到正无穷之间的值,用于限制每个位置可能出现的氨基酸")
argparser.add_argument("--pssm_log_odds_flag", type=int, default=0, help="0 表示 False,1 表示 True")
argparser.add_argument("--pssm_bias_flag", type=int, default=0, help="0 表示 False,1 表示 True")
argparser.add_argument("--tied_positions_jsonl", type=str, default='', help="包含约束位置关系的字典文件路径")
例如,要创建一个用于运行 ProteinMPNN 的 conda 环境:
conda create --name mlfold- 这将创建名为mlfold的 conda 环境source activate mlfold- 激活该环境conda install pytorch torchvision torchaudio cudatoolkit=11.3 -c pytorch- 按照 https://pytorch.org/ 上的步骤安装 PyTorch
以下是在 examples/ 目录中提供的示例脚本:
submit_example_1.sh- 简单的单体示例submit_example_2.sh- 简单的多链示例submit_example_3.sh- 直接从 .pdb 文件路径输入submit_example_3_score_only.sh- 仅返回得分(模型不确定性)submit_example_3_score_only_from_fasta.sh- 仅返回得分(模型不确定性),从 FASTA 文件加载序列submit_example_4.sh- 固定某些残基位置submit_example_4_non_fixed.sh- 指定需要设计的特定位置submit_example_5.sh- 将某些位置绑定在一起(对称性)submit_example_6.sh- 同源寡聚体示例submit_example_7.sh- 返回序列的无条件概率(类似 PSSM)submit_example_8.sh- 添加氨基酸偏好submit_example_pssm.sh- 在序列设计时使用 PSSM 偏好
输出示例:
>3HTN, score=1.1705, global_score=1.2045, fixed_chains=['B'], designed_chains=['A', 'C'], model_name=v_48_020, git_hash=015ff820b9b5741ead6ba6795258f35a9c15e94b, seed=37
NMYSYKKIGNKYIVSINNHTEIVKALNAFCKEKGILSGSINGIGAIGELTLRFFNPKTKAYDDKTFREQMEISNLTGNISSMNEQVYLHLHITVGRSDYSALAGHLLSAIQNGAGEFVVEDYSERISRTYNPDLGLNIYDFER/NMYSYKKIGNKYIVSINNHTEIVKALNAFCKEKGILSGSINGIGAIGELTLRFFNPKTKAYDDKTFREQMEISNLTGNISSMNEQVYLHLHITVGRSDYSALAGHLLSAIQNGAGEFVVEDYSERISRTYNPDLGLNIYDFER
>T=0.1, sample=1, score=0.7291, global_score=0.9330, seq_recovery=0.5736
NMYSYKKIGNKYIVSINNHTEIVKALKKFCEEKNIKSGSVNGIGSIGSVTLKFYNLETKEEELKTFNANFEISNLTGFISSMHDNKVFLDLHITIGDENFSALAGHLLSAVVNGTCELIVEDFNELVSTKYNEELGLWLLDFEK/NMYSYKKIGNKYIVSINNHTDIVTAIKKFCEDKKIKSGTINGIGQVKEVTLEFRNFETGEKEEKTFKKQFTISNLTGFISTKDGKVFLDLHITFGDENFSALAGHLLSAIVDGKCELIIEDYNEEINVKYNEELGLYLLDFNK
>T=0.1, sample=2, score=0.7414, global_score=0.9355, seq_recovery=0.6075
NMYKYKKIGNKYIVSINNHTEIVKAIKEFCKEKNIKSGTINGIGQVGKVTLRFYNPETKEYTEKTFNDNFEISNLTGFISTYKNEVFLHLHITFGKSDFSALAGHLLSAIVNGICELIVEDFKENLSMKYDEKTGLYLLDFEK/NMYKYKKIGNKYVVSINNHTEIVEALKAFCEDKKIKSGTVNGIGQVSKVTLKFFNIETKESKEKTFNKNFEISNLTGFISEINGEVFLHLHITIGDENFSALAGHLLSAVVNGEAILIVEDYKEKVNRKYNEELGLNLLDFNL
score- 对所有被设计的残基,采样到的氨基酸的负对数概率的平均值global score- 对所有链中的所有残基,采样或固定的氨基酸的负对数概率的平均值fixed_chains- 未被设计、保持固定的链designed_chains- 被重新设计的链model_name/CA_model_name- 用于生成结果的模型名称,例如v_48_020git_hash- 用于生成输出的 GitHub 版本号seed- 随机种子T=0.1- 使用温度 0.1 来采样序列sample- 序列样本编号,如 1、2、3 等
@article{dauparas2022robust,
title={Robust deep learning--based protein sequence design using ProteinMPNN},
author={Dauparas, Justas and Anishchenko, Ivan and Bennett, Nathaniel and Bai, Hua and Ragotte, Robert J and Milles, Lukas F and Wicky, Basile IM and Courbet, Alexis and de Haas, Rob J and Bethel, Neville and others},
journal={Science},
volume={378},
number={6615},
pages={49--56},
year={2022},
publisher={American Association for the Advancement of Science}
}
版本历史
v1.0.12022/08/19v1.0.02022/07/29常见问题
相似工具推荐
ML-For-Beginners
ML-For-Beginners 是由微软推出的一套系统化机器学习入门课程,旨在帮助零基础用户轻松掌握经典机器学习知识。这套课程将学习路径规划为 12 周,包含 26 节精炼课程和 52 道配套测验,内容涵盖从基础概念到实际应用的完整流程,有效解决了初学者面对庞大知识体系时无从下手、缺乏结构化指导的痛点。 无论是希望转型的开发者、需要补充算法背景的研究人员,还是对人工智能充满好奇的普通爱好者,都能从中受益。课程不仅提供了清晰的理论讲解,还强调动手实践,让用户在循序渐进中建立扎实的技能基础。其独特的亮点在于强大的多语言支持,通过自动化机制提供了包括简体中文在内的 50 多种语言版本,极大地降低了全球不同背景用户的学习门槛。此外,项目采用开源协作模式,社区活跃且内容持续更新,确保学习者能获取前沿且准确的技术资讯。如果你正寻找一条清晰、友好且专业的机器学习入门之路,ML-For-Beginners 将是理想的起点。
ragflow
RAGFlow 是一款领先的开源检索增强生成(RAG)引擎,旨在为大语言模型构建更精准、可靠的上下文层。它巧妙地将前沿的 RAG 技术与智能体(Agent)能力相结合,不仅支持从各类文档中高效提取知识,还能让模型基于这些知识进行逻辑推理和任务执行。 在大模型应用中,幻觉问题和知识滞后是常见痛点。RAGFlow 通过深度解析复杂文档结构(如表格、图表及混合排版),显著提升了信息检索的准确度,从而有效减少模型“胡编乱造”的现象,确保回答既有据可依又具备时效性。其内置的智能体机制更进一步,使系统不仅能回答问题,还能自主规划步骤解决复杂问题。 这款工具特别适合开发者、企业技术团队以及 AI 研究人员使用。无论是希望快速搭建私有知识库问答系统,还是致力于探索大模型在垂直领域落地的创新者,都能从中受益。RAGFlow 提供了可视化的工作流编排界面和灵活的 API 接口,既降低了非算法背景用户的上手门槛,也满足了专业开发者对系统深度定制的需求。作为基于 Apache 2.0 协议开源的项目,它正成为连接通用大模型与行业专有知识之间的重要桥梁。
PaddleOCR
PaddleOCR 是一款基于百度飞桨框架开发的高性能开源光学字符识别工具包。它的核心能力是将图片、PDF 等文档中的文字提取出来,转换成计算机可读取的结构化数据,让机器真正“看懂”图文内容。 面对海量纸质或电子文档,PaddleOCR 解决了人工录入效率低、数字化成本高的问题。尤其在人工智能领域,它扮演着连接图像与大型语言模型(LLM)的桥梁角色,能将视觉信息直接转化为文本输入,助力智能问答、文档分析等应用场景落地。 PaddleOCR 适合开发者、算法研究人员以及有文档自动化需求的普通用户。其技术优势十分明显:不仅支持全球 100 多种语言的识别,还能在 Windows、Linux、macOS 等多个系统上运行,并灵活适配 CPU、GPU、NPU 等各类硬件。作为一个轻量级且社区活跃的开源项目,PaddleOCR 既能满足快速集成的需求,也能支撑前沿的视觉语言研究,是处理文字识别任务的理想选择。
awesome-machine-learning
awesome-machine-learning 是一份精心整理的机器学习资源清单,汇集了全球优秀的机器学习框架、库和软件工具。面对机器学习领域技术迭代快、资源分散且难以甄选的痛点,这份清单按编程语言(如 Python、C++、Go 等)和应用场景(如计算机视觉、自然语言处理、深度学习等)进行了系统化分类,帮助使用者快速定位高质量项目。 它特别适合开发者、数据科学家及研究人员使用。无论是初学者寻找入门库,还是资深工程师对比不同语言的技术选型,都能从中获得极具价值的参考。此外,清单还延伸提供了免费书籍、在线课程、行业会议、技术博客及线下聚会等丰富资源,构建了从学习到实践的全链路支持体系。 其独特亮点在于严格的维护标准:明确标记已停止维护或长期未更新的项目,确保推荐内容的时效性与可靠性。作为机器学习领域的“导航图”,awesome-machine-learning 以开源协作的方式持续更新,旨在降低技术探索门槛,让每一位从业者都能高效地站在巨人的肩膀上创新。
scikit-learn
scikit-learn 是一个基于 Python 构建的开源机器学习库,依托于 SciPy、NumPy 等科学计算生态,旨在让机器学习变得简单高效。它提供了一套统一且简洁的接口,涵盖了从数据预处理、特征工程到模型训练、评估及选择的全流程工具,内置了包括线性回归、支持向量机、随机森林、聚类等在内的丰富经典算法。 对于希望快速验证想法或构建原型的数据科学家、研究人员以及 Python 开发者而言,scikit-learn 是不可或缺的基础设施。它有效解决了机器学习入门门槛高、算法实现复杂以及不同模型间调用方式不统一的痛点,让用户无需重复造轮子,只需几行代码即可调用成熟的算法解决分类、回归、聚类等实际问题。 其核心技术亮点在于高度一致的 API 设计风格,所有估算器(Estimator)均遵循相同的调用逻辑,极大地降低了学习成本并提升了代码的可读性与可维护性。此外,它还提供了强大的模型选择与评估工具,如交叉验证和网格搜索,帮助用户系统地优化模型性能。作为一个由全球志愿者共同维护的成熟项目,scikit-learn 以其稳定性、详尽的文档和活跃的社区支持,成为连接理论学习与工业级应用的最
keras
Keras 是一个专为人类设计的深度学习框架,旨在让构建和训练神经网络变得简单直观。它解决了开发者在不同深度学习后端之间切换困难、模型开发效率低以及难以兼顾调试便捷性与运行性能的痛点。 无论是刚入门的学生、专注算法的研究人员,还是需要快速落地产品的工程师,都能通过 Keras 轻松上手。它支持计算机视觉、自然语言处理、音频分析及时间序列预测等多种任务。 Keras 3 的核心亮点在于其独特的“多后端”架构。用户只需编写一套代码,即可灵活选择 TensorFlow、JAX、PyTorch 或 OpenVINO 作为底层运行引擎。这一特性不仅保留了 Keras 一贯的高层易用性,还允许开发者根据需求自由选择:利用 JAX 或 PyTorch 的即时执行模式进行高效调试,或切换至速度最快的后端以获得最高 350% 的性能提升。此外,Keras 具备强大的扩展能力,能无缝从本地笔记本电脑扩展至大规模 GPU 或 TPU 集群,是连接原型开发与生产部署的理想桥梁。